Evaluate principles to increase sampling precision in experimental design;
Evaluate power of sampling;
Demonstrate power tools in R.
2 Start a Script
For this lab or project, begin by:
Starting a new R script
Create a good header section and table of contents
Save the script file with an informative name
set your working directory
Aim to make the script a future reference for doing things in R!
3 Introduction
Before carrying out a time- and fund-consuming experiment, it is useful to get an idea of what to expect from the results. How big an effect are you expecting? What are the chances that you would detect it? What sample size would you need to have a reasonable chance of succeeding? How narrow a confidence interval around the estimated effect would you be happy with? In this lab we will show how R can be used to address some of these questions.
4 Random Sampling
To begin, let’s get some practice sampling (randomly) categorical and Gaussian-distributed data from a population. The intention is to use the sample() function. The sample() function has four arguments:
x: a vector of values to sample from;
size: the number of samples to take;
replace: whether to sample with replacement (TRUE) or without replacement (FALSE);
prob: a vector of probabilities for each value in x (only used if replace = TRUE).
Let’s use the sample() function to:
Randomly sample 20 observations from a population having two groups of individuals, “infected” and “uninfected”, in equal proportions. Summarize the results in a frequency table;
Repeat the previous step five times to convince yourself that the outcome varies from sample to sample;
Sample 18 individuals from a population having two groups of individuals, “mated” and “unmated”, where the proportion mated in the population is 0.7. Summarize the results in a frequency table;
Repeat the previous step five times to convince yourself that the outcome varies from sample to sample;
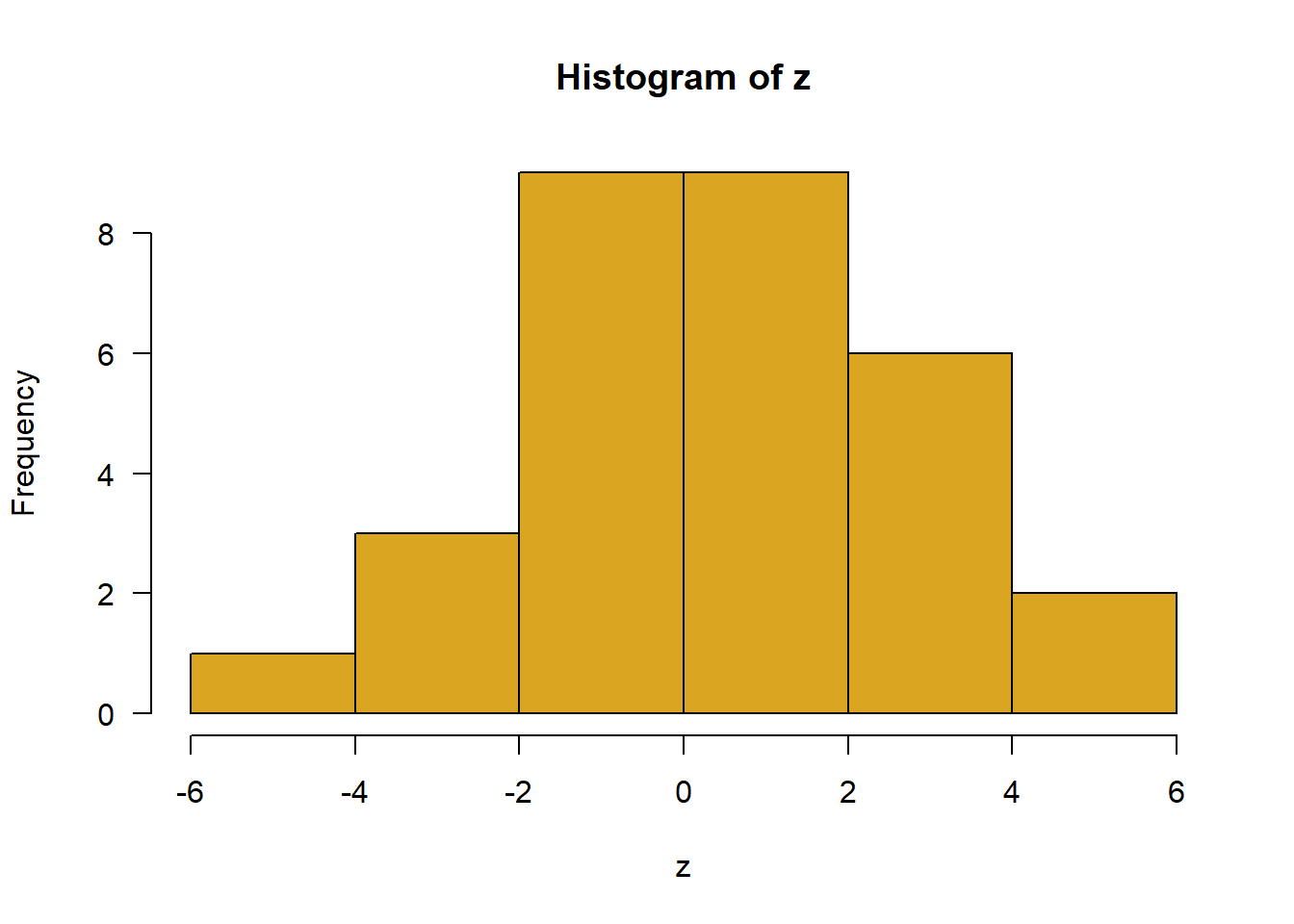
Sample 30 observations from a Gaussian-distributed population having mean 0 and standard deviation 2. Plot the results in a histogram;
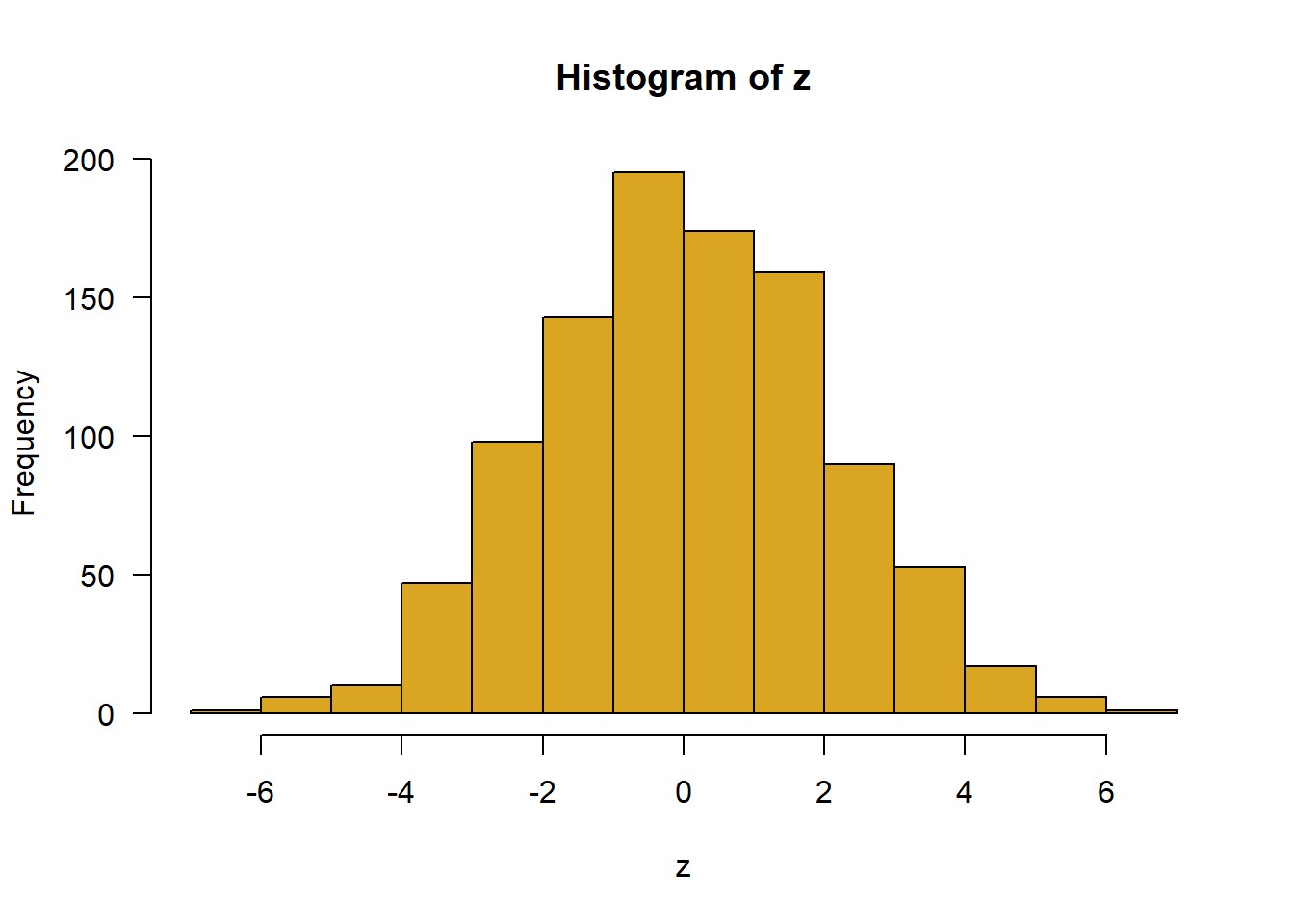
Repeat the following 5 times and calculate the mean each time: sample 30 observations from a Gaussian-distributed population having mean 0 and standard deviation 2. Convince yourself that the sample mean is different each time.
# 1. run this several times in your own script and see how the output changessample(c("infected","uninfected"), prob =c(.5,.5),size =20, replace =TRUE)
# 3. z <-sample(c("mated","unmated"), size =18, replace =TRUE, prob =c(.7,.3))table(z)
z
mated unmated
14 4
# 5. z <-rnorm(30, mean =0, sd =2)hist(z, right =FALSE, col ="goldenrod", las =1)
# 6. z <-rnorm(1000, mean =0, sd =2)hist(z, right =FALSE, col ="goldenrod", las =1)
5 Plan for Precision
Consider an experiment to estimate mate preference of females of a species of jumping spider. Each independent trial involves presenting a female spider with two tethered males. One of the males is from her own species, and the other is from its sister species. To avoid pseudoreplication (i.e., sampling the same observation more than once), females are tested only once and males are replaced between tests. You want to estimate p, the proportion of female spiders that choose males of their own species. Before carrying out the experiment, it is useful to generate data under different scenarios to get a sense of the sample size you would need to estimate preference with sufficient precision.
5.1 Weak or no preference
We’ll start with the case of weak or no preference: imagine that females choose males essentially randomly (p = 0.5), with half choosing the male from her own species and the other half picking the male of the other species. How much data would you need to demonstrate this (and convince your skeptical supervisory committee)? One idea is to collect data and use it to test the null hypothesis of no preference. If the null hypothesis is true, you should fail to reject it. However, this won’t be very convincing. Failing to reject a null hypothesis is inconclusive by itself. Maybe your test won’t have much power.
A second idea is to plan your sample size so as to obtain a narrow confidence interval (i.e., having high precision) for the strength of preference. If, at the end of your experiment, you end up with an estimate of p close to 0.5 AND your 95% confidence interval for p is relatively narrow, you’ll be in a strong position to say that the true preference really is weak, even if you can’t say it is exactly 0.5. What sample size is necessary to achieve a reasonably narrow confidence interval in this case? We can investigate this question by simulating data:
We will randomly sample n = 10 females from a population having equal numbers of “successes” (females who choose males of her own species) and “failures” (females who choose males of the other species):
# 10 females from a population having equal numbers # of successes (1) and failures (0)x <-sample(c(1, 0), size =10, c(0.5, 0.5), replace =TRUE)# View samplex
[1] 1 1 0 1 0 0 0 1 1 1
# Calculate proportion of successessum(x)/10
[1] 0.6
Using this data we will calculate an approximate 95% confidence interval for the population proportion of successes. We can use the Agresti-Coull method in the binom package. To obtain the 95% confidence interval, use the binom.confint function explained below. The argument x is the number of “successes” in your generated sample (number of females who chose males of her own species) and n is the sample size (number of females tested):
# if(!require("binom")) install.packages("binom")# Load the packagelibrary(binom)# Get the confidence intervaln <-50# number of trialsx <-27# number of successesmyCI <-binom.confint(x, n, method ="ac") print(myCI) # shows the results
method x n mean lower upper
1 agresti-coull 27 50 0.54 0.4039603 0.6703319
myCI$lower # lower limit of confidence interval
[1] 0.4039603
myCI$upper # upper limit
[1] 0.6703319
Use this code to do the following:
Obtain the 95% confidence interval for the proportion using your data from step 1. What was the span of your confidence interval (upper limit minus lower limit)? Can you be confident that the true population proportion is 0.5 or very close to it?
Repeat steps 1 and 2 five times and keep a record of the confidence intervals you obtained. What was the lowest value for the span of the confidence interval from the 5 samples?
You can speed up the effort if you create a for loop in R that automatically repeats steps 1 and 2 as many times as you decide. A loop that repeats ten times would look something like the following. The “i” in this loop is a counter, starting at 1 and increasing by 1 each time the commands in the loop are executed. Don’t forget to include a command inside the loop to print each result.
Use this code as a template for your for loop:
for(i in1:10) {# [paste in your R commands for steps 1 and 2 here]}
Increase the sample size to n = 20 and run the loop from step 4 again. How much narrower are the confidence interval spans? Are the spans adequate?
By modifying the sample size and re-running the loop a bunch of times, find a sample size (ballpark, no need to be exact at this point) that usually produces a confidence interval having a span no greater than 0.2. This would be the span of a confidence interval that had, e.g., a lower limit of 0.4 and an upper limit of 0.6. Surely this would be convincing evidence that the mate preference really was weak.
By this point you might wish to speed things up by saving the results of each iteration to a vector or data frame rather than print the results to the screen. This will make it possible to increase the number of iterations (say, to 100 times instead of just 10) for a given value of n.
Given the results of step 6, you would now have some design options before you. Is the sample size n that your simulation indicated was needed to generate a confidence interval of span 0.2 realistic? In other words, would an experiment with so many female spiders (and so many males) be feasible? If the answer is yes, great, get started on your experiment! If the answer is no, the sample size required is unrealistically large, then you have some decisions to make. Here are some options:
Forget all about doing the experiment (consider a thesis based on theory instead);
Revise your concept of what represents a “narrow” confidence interval. Maybe a confidence interval for p spanning, say, 0.3 to 0.7 (a span of 0.4) would be good enough to allow you to conclude that the preference was “not strong”. This would not require as big a sample size as a narrower interval.
Repeat the above procedures to find a sample size that usually gives a confidence interval having a span of 0.4 or less.
💡 Click here to view a solution
# Ensure the 'binom' package is installed and loadedif(!require("binom")) install.packages("binom")library(binom, quietly =TRUE, warn.conflicts =FALSE)# Function to simulate one trial and calculate the confidence intervalsimulate_trial <-function(n) {# Generate a sample with n females, equal probability of success and failure x <-sample(c(1, 0), size = n, replace =TRUE, prob =c(0.5, 0.5))# Number of successes (females choosing males of their own species) num_successes <-sum(x)# Calculate 95% confidence interval using Agresti-Coull method ci <-binom.confint(num_successes, n, method ="ac")# Return the span of the confidence intervalreturn(ci$upper - ci$lower)}# Function to simulate multiple trials and find the sample size for desired CI spanfind_sample_size <-function(target_span, max_iterations =100, start_n =10, increment =10) {for (n inseq(from = start_n, by = increment, length.out = max_iterations)) {# Simulate 5 trials for each sample size spans <-replicate(5, simulate_trial(n))# Check if any of the spans is less than or equal to the target spanif (any(spans <= target_span)) {return(list(sample_size = n, spans = spans)) } }return(list(sample_size =NA, spans =NA))}# Step 5: Find sample size for a CI span of 0.2result_02 <-find_sample_size(0.2)print(result_02)
Assume that the preference p really is different from 0.5, and use null hypothesis significance testing to detect it. What strength of preference would we like to be able to detect in our experiment? To pick an extreme case, if the true proportion of females in the population choosing a male from her own species is 0.51 rather than 0.50, you would need an enormous sample size to detect it. But we don’t really care about such a small effect. Let’s start instead with the more realistic proportion p = 0.7. What sample size would be needed to detect it with reasonably high probability?
6.1 Binomial test
Let’s start my sampling 20 females from a population in which the true fraction of “successes” is 0.7:
# set the seed so that the results are reproducibleset.seed(99)# Sample 20 observations from a population in which the true fraction of successes is 0.7x <-sample(c("success","failure"), size =20, c(0.7,0.3), replace =TRUE)# Count the number of successesnsuccess <-length(which(x =="success"))
The we can use the binomial test to test the null hypothesis that the true proportion of successes is 0.5. This test calculates the exact 2-tailed probability of a result as extreme or more extreme as that observed if the null hypothesis is true. We can implement it in R using the binom.test() function:
# Calculate the exact 2-tailed probability of a result as extreme or more extreme as that observed if the null hypothesis is truez <-binom.test(nsuccess, 20, 0.5)# Print the resultsprint(z)
Exact binomial test
data: nsuccess and 20
number of successes = 18, number of trials = 20, p-value = 0.0004025
alternative hypothesis: true probability of success is not equal to 0.5
95 percent confidence interval:
0.6830173 0.9876515
sample estimates:
probability of success
0.9
# Print the p-valuez$p.value
[1] 0.0004024506
In this function x is the observed number of successes in your sample from step 1, and n is the sample size. z here is an object that stores the result. Based on the p-value we can reject the null hypothesis that the true proportion of successes is 0.5. But what if we had sampled 20 individuals from a population in which the true fraction of successes is 0.6? Would we have been able to reject the null hypothesis? Let’s find out by repeating the above steps 10 times:
# Set the seed so that the results are reproducibleset.seed(99)# Create a vector to store the resultsp <-rep(NA, 10)# Repeat the following 10 timesfor(i in1:10){# Sample 20 observations from a population in which the true fraction of successes is 0.6 x <-sample(c("success","failure"), size =20, c(0.6,0.4), replace =TRUE)# Count the number of successes nsuccess <-length(which(x =="success"))# Calculate the exact 2-tailed probability of a result as extreme or more extreme as that observed if the null hypothesis is true z <-binom.test(nsuccess, 20, 0.5)# Store the p-value p[i] <- z$p.value}
We can use this to determine what fraction of iterations the null hypothesis was rejected:
# Print the number of times the null hypothesis was rejectedlength(which(p <0.05))
[1] 1
# Print the fraction of times the null hypothesis was rejectedlength(which(p <0.05)) /10
[1] 0.1
Let’s modifying the sample size and re-run the loop multiple times to find a sample size (ballpark, no need to be exact at this point) that usually results in the null hypothesis being rejected:
# Set the seed so that the results are reproducibleset.seed(99)# Create a vector to store the resultsp <-rep(NA, 2000)# Repeat the following 2000 timesfor(i in1:2000){# Sample 50 observations from a population in which the true fraction of successes is 0.6 x <-sample(c("success","failure"), size =50, c(0.6,0.4), replace =TRUE)# Count the number of successes nsuccess <-length(which(x =="success"))# Calculate the exact 2-tailed probability of a result as extreme or more extreme as that observed if the null hypothesis is true z <-binom.test(nsuccess, 50, 0.5)# Store the p-value p[i] <- z$p.value}# Print the fraction of times the null hypothesis was rejectedlength(which(p <0.05)) /10
[1] 48.6
A sample size of 2000 is liekly too large to be feasible in an experiment - you have some experimental design decisions to make. ou could decide not to run the experiment after all. Or, you could revise your aims. Perhaps you could detect a preference of 0.7 instead of 0.6?
6.2 Power tools in R
Simulating random samples on the computer, as we did above, is a great way to investigate power and sample size requirements. It works in any situation and can mimic, even complicated study designs. However, a number of quantitative tools have been developed for mainly simple designs that do the work for you.
Load the pwr package and we will look at how it can do some of the calculations for you. Let’s use this package to calculate the approximate minimum sample size needed to detect a preference of 0.6 with a power of 0.80 (i.e., the null hypothesis would be rejected in 80% of experiments). The null hypothesis is that the population proportion p of females who would choose the male from her own population is 0.5. The goal is to design an experiment that has a high probability of rejecting the null hypothesis when p is 0.6.
# You might need to install {pwr}library(pwr)# Calculate the effect sizeh <-ES.h(0.5, 0.6)# Calculate the minimum sample sizez <-pwr.p.test(h, power =0.8)# Print the resultsprint(z)
proportion power calculation for binomial distribution (arcsine transformation)
h = 0.2013579
n = 193.5839
sig.level = 0.05
power = 0.8
alternative = two.sided
We can also use the this approach to calculate values for a range of preferences:
# Repeat for range values of prefpref <-c(0.6, 0.7, 0.8, 0.9)# Loop through the values in preffor(i in1:length(pref)){ h <-ES.h(0.5, pref[i]) z <-pwr.p.test(h, power =0.8)print(z$n)}
In an experiment on the great tit, two eggs were removed from 30 nests, which caused the attending females to lay one more egg. 35 un-manipulated nests served as controls. The response variable was incidence of malaria in female great tits at the end of the experiment. The results of the experiment are tabulated below:
Control birds
Egg-removal birds
Malaria
7
15
No malaria
28
15
Imagine that you are considering repeating this experiment on a different species of songbird. What are the chances of detecting an effect? What sample sizes should you plan?
Let’s start my randomly sampling 30 females from a control population in which the fraction of infected birds is 0.2 (the fraction in the tit data). We will also sample 30 females from an experimental population in which the fraction of infected birds is 0.5 (the fraction in the tit data). We can combine these samples into a data frame and include a variable indicating treatment:
Let’s look at the 2 x 2 frequency table from your random sample to determine if there an association:
# View frequency tabletable(mydata)
treatment
response control treat
malaria 2 14
no 28 16
We can loop this to repeat the process 5 times to show that these are randomly sampled:
# Function to generate datagenerate_data <-function() {# Control population x <-sample(c("malaria", "no"), size =30, replace =TRUE, prob =c(0.2, 0.8))# Experimental population y <-sample(c("malaria", "no"), size =30, replace =TRUE, prob =c(0.5, 0.5))# Treatment z <-rep(c("control", "treat"), each =30)# Combine into a data framedata.frame(response =c(x, y), treatment = z)}# Loop to generate, create table, and print the data 3 timesfor (i in1:5) {cat("Iteration", i, ":\n")print(table(generate_data()))cat("\n") # Adds an extra line for readability}
Iteration 1 :
treatment
response control treat
malaria 3 16
no 27 14
Iteration 2 :
treatment
response control treat
malaria 5 20
no 25 10
Iteration 3 :
treatment
response control treat
malaria 4 9
no 26 21
Iteration 4 :
treatment
response control treat
malaria 7 19
no 23 11
Iteration 5 :
treatment
response control treat
malaria 5 17
no 25 13
Let’s use the tools in pwr to calculate the sample size needed to achieve 80% power in this design:
# Load the pwr packagelibrary(pwr)# Define control and treatment proportionscontrol <-c(0.2,0.8)treatment <-c(0.5,0.5)# Combine into a data frameprobs <-cbind.data.frame(treatment = treatment,control = control,stringsAsFactors =FALSE)# Cohen's effect size "w"w <-ES.w2(probs/sum(probs)) # Calculate the sample sizez <-pwr.chisq.test(w, df =1, power =0.80)z$N
[1] 79.36072
8 Plan a 2-Treatment Experiment
Imagine that you are considering a two-treatment experiment for a numeric response variable. The treatments consist of two grazing regimes and the response variable is the number of plant species in plots at the end of the experiment. How many replicate plots should you set up? As usual, we will investigate only the case of equal sample sizes in the two groups. We’ll assume that the number of plant species in plots has a Gaussian (i.e., normal) distribution in both treatment and control. We’ll round the numbers so that they are integers.
Let’s start by randomly sampling 20 measurements from a Gaussian distribution having a mean of 10 and a variance of 10 (so the standard deviation is the square root of 10). Call this the “control” group:
# Randomly sample 20 measurements from a Gaussian distribution# having a mean of 10 and a variance of 10control <-rnorm(20, mean =10, sd =sqrt(10))# Round the numbers so that they are integerscontrol <-round(control, 0)# View the datacontrol
Let’s repeat this for a second sample. We will sample from a Gaussian distribution having a mean of 15 but the same sample variance, 10. (This is a bit unrealistic, as we would expect the variance in numbers of species to be higher as the mean increases, but never mind for now). Call this the “treatment” group. In other words, we will investigate the power of this experiment to detect a 1.5-fold change in the mean number of species from control to treatment.
# Randomly sample 20 measurements from a Gaussian distribution# having a mean of 15 and a variance of 10treatment <-rnorm(20, mean =15, sd =sqrt(10))# Round the numbers so that they are integerstreatment <-round(treatment, 0)# View the datatreatment
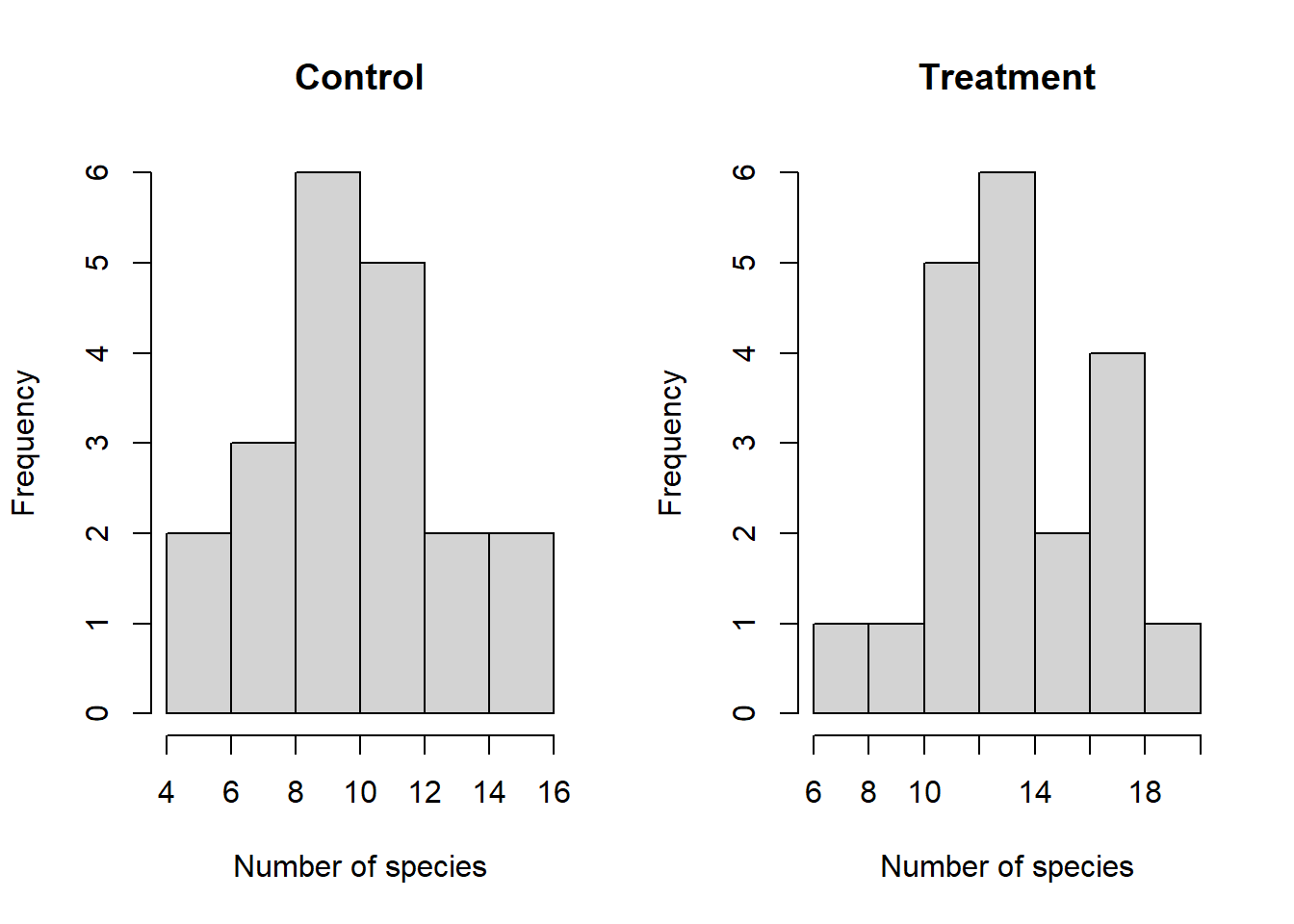
We need to assemble the samples into a data frame in “long” format, with a second variable indicating which measurements are from the control group and which are from the treatment group. Create a histogram using base R for each of the two samples and compare the distributions by eye:
# Combine the samples into a data framemydata <-cbind.data.frame(response =c(control, treatment),treatment =rep(c("control", "treat"), each =20),stringsAsFactors =FALSE)# Set graph parameterspar(mfrow =c(1, 2)) # Set up a 1 x 2 plotting space# Histogram of control grouphist(mydata$response[mydata$treatment =="control"],main ="Control",xlab ="Number of species")# Histogram of treatment grouphist(mydata$response[mydata$treatment =="treat"],main ="Treatment",xlab ="Number of species")
We can use the power.t.test() function to determine the power of the above design – probability that the experiment will detect a significant difference between the treatment and control means based on random samples:
# Calculate the power of the experimentpower.t.test(n =20, delta =1.5, sd =sqrt(10), sig.level =0.05, type ="two.sample")
Two-sample t test power calculation
n = 20
delta = 1.5
sd = 3.162278
sig.level = 0.05
power = 0.3093157
alternative = two.sided
NOTE: n is number in *each* group
This only gives us a power of 0.31, which is not very good. Let’s use the same command to determine the sample size that would be necessary to achieve a power of 0.80:
# Calculate the sample size needed to achieve a power of 0.80power.t.test(delta =1.5, sd =sqrt(10), sig.level =0.05, power =0.80, type ="two.sample")
Two-sample t test power calculation
n = 70.74111
delta = 1.5
sd = 3.162278
sig.level = 0.05
power = 0.8
alternative = two.sided
NOTE: n is number in *each* group
We would need a sample size of 70 in each group to achieve a power of 0.80.