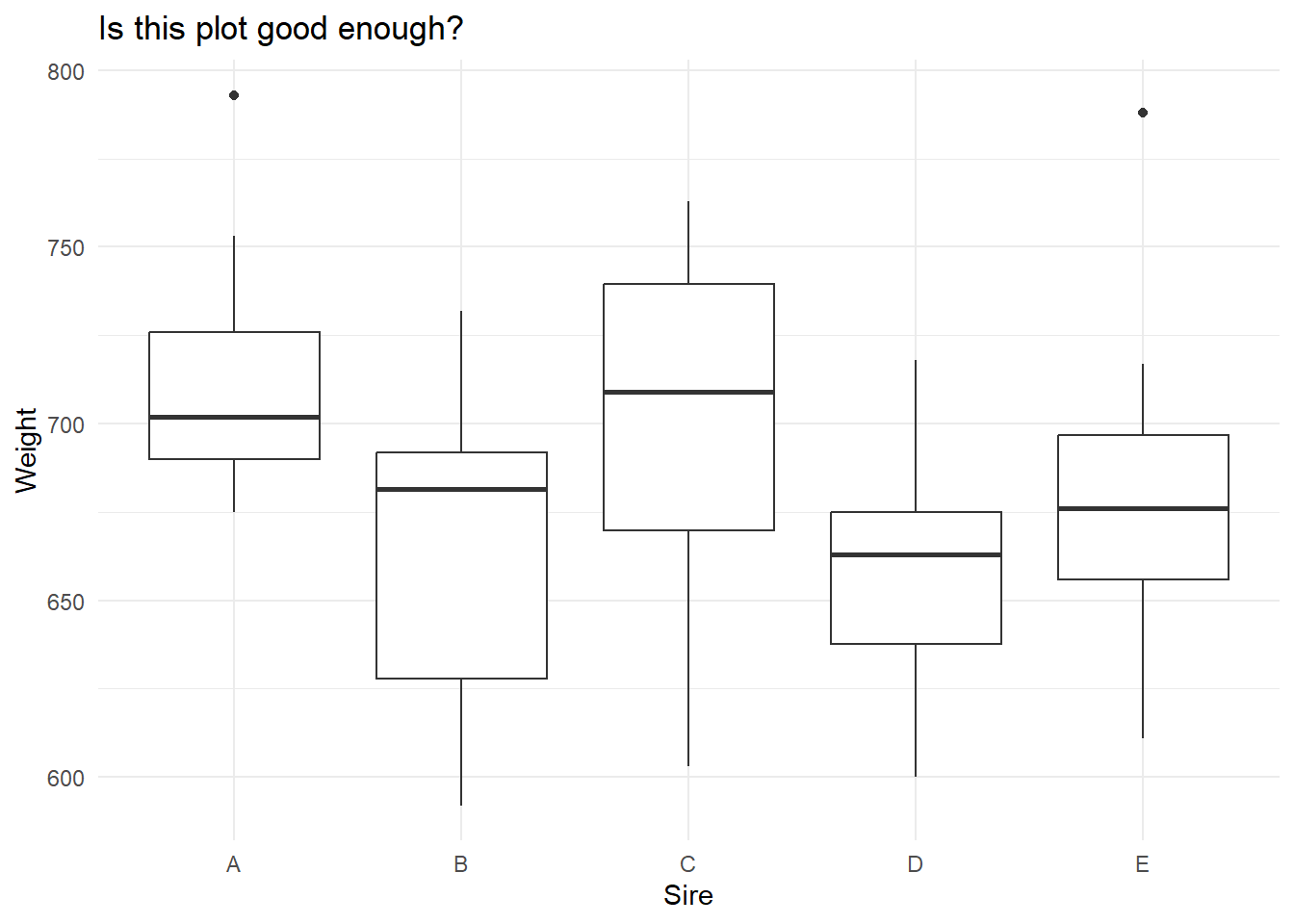Use R to perform analysis of variance (ANOVA) to compare the means of multiple groups;
Perform Tukey-Kramer tests to look at unplanned contrasts between all pairs of groups;
Use Kruskal-Wallis tests to test for difference between groups without assumptions of Gaussianity;
Transform data to meet assumptions of ANOVA.
2 Start a Script
For this lab or project, begin by:
Starting a new R script
Create a good header section and table of contents
Save the script file with an informative name
set your working directory
Aim to make the script a future reference for doing things in R!
3 Introduction
Perhaps more so than any other tool, the Analysis of Variance (ANOVA) played a role in literally revolutionising the idea of objectivity in using data to produce evidence to support claims for certain experimental designs. Invented by the famous statistician and biologist R. A. Fisher while he worked at Rothamsted Research, the intention was for ANOVA to be a useful tool to analyse agriculture experiments. Today, despite many innovations and competing approaches, it remains at the foundation of the basic practice of statistics.
4 Why ANOVA?
Two-sample t-tests evaluate whether or not the mean of a numeric variable changes among two groups or experimental conditions, which can be encoded by a categorical variable. We can conceptualise this test as one that evaluates whether or not the numeric variable is related to the categorical variable, reflected as a difference between means of different groups. What happens if we need to evaluate differences among means of more than two groups? The ‘obvious’ thing to do might seem to be to test each pair of means using a t-test. However, this procedure is statistically flawed. The problem is that the more tests we do, the more likely we are to make a Type I error (i.e., a false positive). This is known as the problem of multiple comparisons. The ANOVA is a statistical test that allows us to evaluate whether or not there is a difference among means of more than two groups, while controlling for the problem of multiple comparisons.
There are several reasons to use a “1-way ANOVA” experimental design. The scenario usually involves:
One numeric continuous dependent variable of interest;
A factor that contains 2 or more levels, often with a control;
When there are just two levels, the 1-ANOVA is conceptually equivalent to the t-test.
An example might be something like a classic field trial, where crop pest damage is measured (the numeric continuous dependent variable) and the factor compares pest treatment with 3 levels: a control level (no pesticide), an organic pesticide, and a chemical pesticide. The basic question is whether there is an overall difference in the numeric dependent variable amongst the factor levels, however several kinds of questions are also possible to answer:
Overall difference test of means between the factors;
Comparison of difference of each factor level with the control or other reference factor level;
Post-hoc tests of difference between specific factor levels, e.g. pairwise tests for a factor with levels A, B, and C might test all possible comparisons A:B, A:C, and B:C;
Examination of the “sources of variation” observed in the dependent variable, e.g. what proportion of total variation can be accounted for by the factor.
The test statistic for ANOVA is the F ratio, which is proportion of variance in the dependent variable between the groups, relative to that within the categories. We will (very briefly) look at this calculation with the aim of gaining a practical understanding of what is going. However, the main focus of this lab is on how to use the R function aov() to perform ANOVA and how to interpret the output.
5 ANOVA Assumptions
The ANOVA is a parametric test, which means that it makes assumptions about the data. The key assumptions for you to be aware of are:
Independence of observations - the groups being compared should be independent of each other. This means that the data collected in one group should not influence the data collected in another. In experimental designs, this is often achieved through random assignment;
Normality - the distribution of the residuals (differences between observed and predicted values) should be approximately normally distributed for each group. This assumption is particularly important when the sample sizes are small. For larger sample sizes, the Central Limit Theorem suggests that this assumption can be somewhat relaxed due to the normalizing effect of large sample sizes;
Homogeneity of variances (homoscedasticity) - the variances among the groups should be approximately equal. This assumption, known as homoscedasticity, ensures that each group contributes equally to the overall analysis. The Levene’s test or Bartlett’s test can be used to check this assumption;
scale of measurement- the dependent variable should be measured at least at the interval level (i.e., the data should be quantitative and continuous). Ratio-level data also meet this criterion;
Random sampling - the data should be collected from a randomly selected portion of the total population. Random sampling helps to generalize the findings from the sample to the population.
5.1 Testing ANOVA assumptions
Let’s see how we can check these assumptions using some example data. The data we will look at is an experiment in animal genetics, looking at the weight of male chickens (8-week old weight in grams), where weight is the continuous dependent variable. The factor is the sire identity, where the measure young male chicks were sired by one of 5 sires, thus sire is a factor with 5 levels A, B, C, D, and E. The data are in wide format, where each column is a different sire and each row is a different chick. We will convert the data to long format, where each row is a different chick and there is a column for the sire identity. This is the format that we need for the ANOVA.
# Load package for data wrangling library(reshape2)
Warning: package 'reshape2' was built under R version 4.3.2
# Create a data frame in wide formatdata_wide <-data.frame(A =c(687, 691, 793, 675, 700, 753, 704, 717),B =c(618, 680, 592, 683, 631, 691, 694, 732),C =c(618, 687, 763, 747, 687, 737, 731, 603),D =c(600, 657, 669, 606, 718, 693, 669, 648),E =c(717, 658, 674, 611, 678, 788, 650, 690))# Convert to long format and rename columnsdata_long <-melt(data_wide, variable.name ="Sire", value.name ="Weight")
No id variables; using all as measure variables
# View the long format data with new column nameshead(data_long)
Sire Weight
1 A 687
2 A 691
3 A 793
4 A 675
5 A 700
6 A 753
To ensure that your data meet the key assumptions for conducting a one-way ANOVA, you can perform several tests in R. To check for normality, you can use the Shapiro-Wilk test, which is available in the stats package. To check for homogeneity of variances, you can use the Levene’s test, which is available in the car package. Remember - we have to check the residuals but to do this we have to fit the model first. We will use the aov() function to fit the model:
Warning: package 'car' was built under R version 4.3.2
Loading required package: carData
Warning: package 'carData' was built under R version 4.3.2
library(ggplot2)# Fit the modelmodel <-aov(Weight ~ Sire, data = data_long)# Check the residualsresiduals <-residuals(model)# Check for normality using the Shapiro-Wilk testshapiro.test(residuals)
Shapiro-Wilk normality test
data: residuals
W = 0.99182, p-value = 0.9913
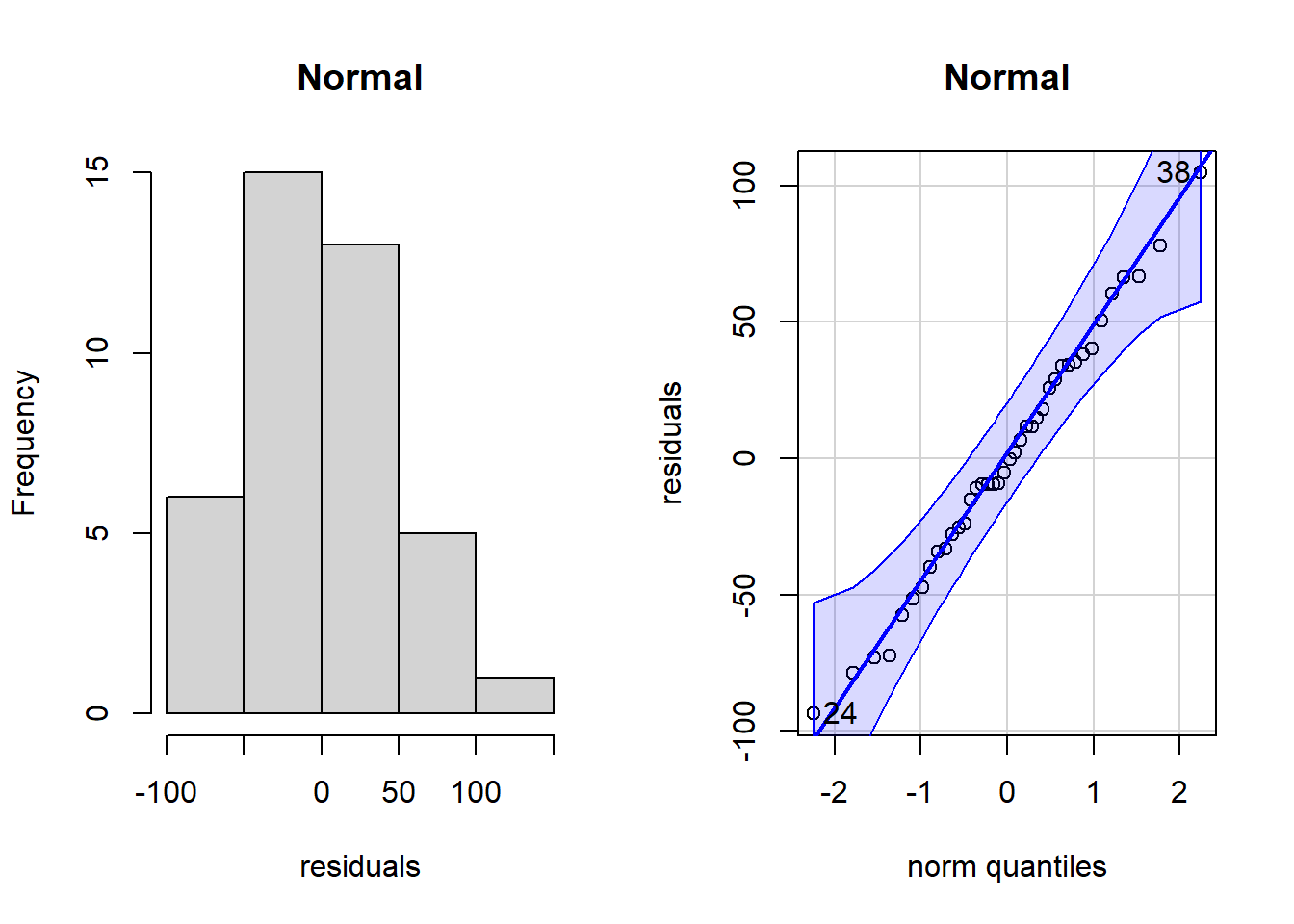
# Graph to examine normality assumption of residualspar(mfrow =c(1,2))hist(residuals,main ="Normal")# Look at residuals with qqPlot()qqPlot(residuals,main ="Normal")
[1] 38 24
At a glance, there are no serious issues with the assumption of normal residual distribution. But we can test for this more formally with a Shapiro-Wilk test:
# Shapiro-Wilk test for normalityshapiro.test(residuals)
Shapiro-Wilk normality test
data: residuals
W = 0.99182, p-value = 0.9913
The null hypothesis of the Shapiro-Wilk test is that the data are normally distributed. There is no evidence of difference to Gaussian in our residuals for our ANOVA model (Shapiro-Wilk: W = 0.99, n = 40, P = 0.99). We can also check for homogeneity of variances using the Bartlett test:
# Levene's test for homogeneity of variancesbartlett.test(Weight ~ Sire, data = data_long)
Bartlett test of homogeneity of variances
data: Weight by Sire
Bartlett's K-squared = 1.6868, df = 4, p-value = 0.7931
The null hypothesis of the Bartlett test is that the variances are equal across groups. We find no evidence that variance in offspring weight differs between sires (Bartlett test: K-sqared = 1.69, df = 4, P = 0.79).
6 Visualising ANOVA
The classic way to visualise the one-way ANOVA is with boxplot, with some way to show the central tendency of the data separately for each factor level. For continuous variables, boxplots show this perfectly. For count variables, barplots are sometimes used with the height set to the mean, along with some form of error bar. Here we will use a boxplot.
# Basic boxplotggplot(data_long, aes(x = Sire, y = Weight)) +geom_boxplot() +ggtitle("Is this plot good enough?") +xlab("Sire") +ylab("Weight") +theme_minimal()
So, it looks like sire identity could have an effect on mean male offspring weight. But, is this graph good enough? Can we make it better? Let’s critique it:
The y axis does not indicate the unit of measurement (important);
Neither axis title is capitalised (important);
Adding on the points might add interesting detail (optional);
Reference line for control (we do not really have a control here) or of the “grand mean” might be useful (optional).
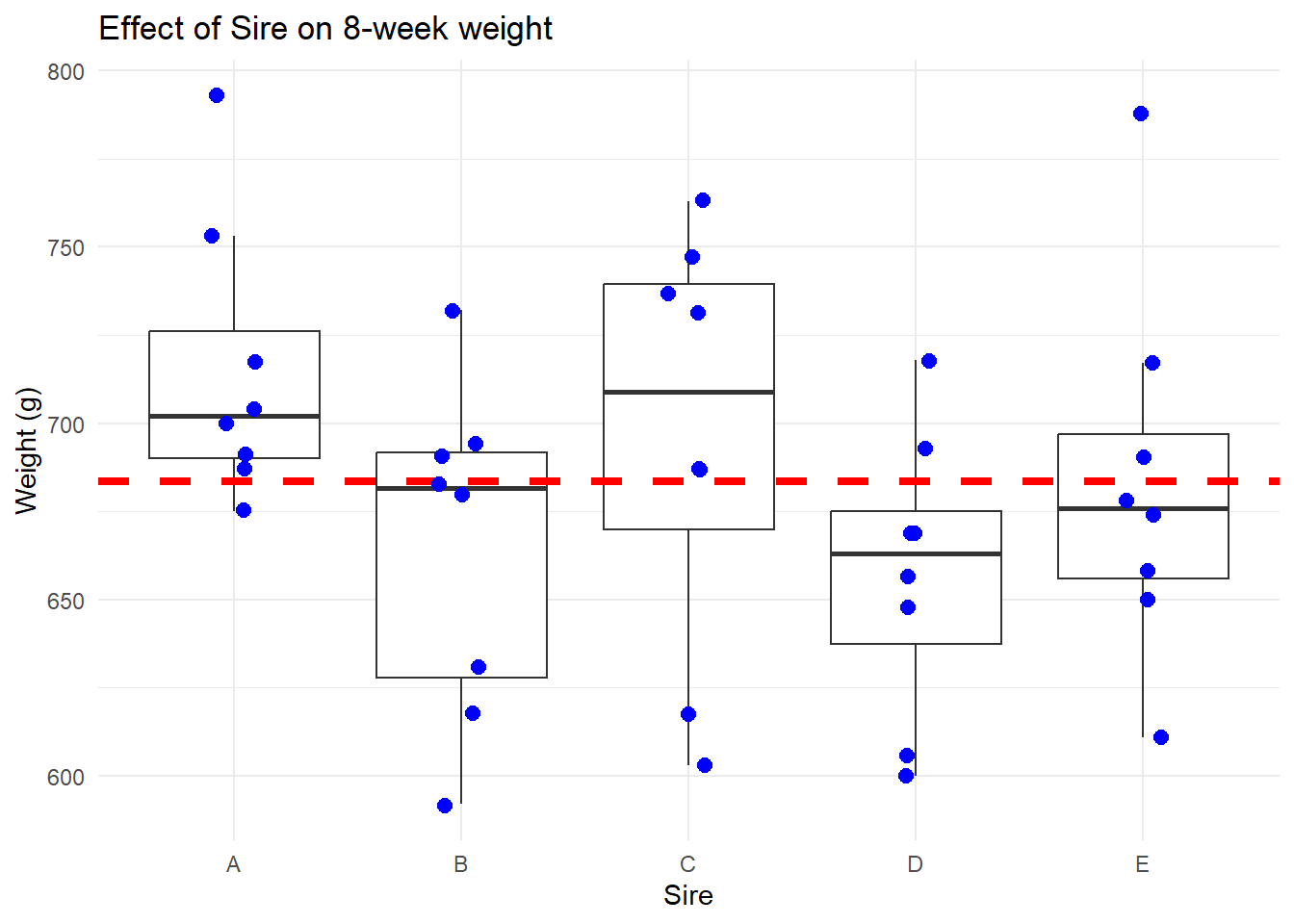
Let’s make a better graph:
# Better boxplotggplot(data_long, aes(x = Sire, y = Weight)) +# Boxplotgeom_boxplot(outlier.shape =NA) +# Exclude outliers for now# Add horizontal line for grand meangeom_hline(yintercept =mean(data_long$Weight), linetype ="dashed", size =1.5, colour ="red") +# Add jittered pointsgeom_jitter(position =position_jitter(0.1), color ="blue", size =2.5) +# Plot labelsggtitle("Effect of Sire on 8-week weight") +xlab("Sire") +ylab("Weight (g)") +# Themetheme_minimal() +theme(legend.position ="none") # Remove legend if not needed
Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
ℹ Please use `linewidth` instead.
7 ANOVA in R
The basic application of ANOVA in R is the aov() function. There are actually a lot of alternative ways to perform the exact same test in R. We will look at few things in this section:
Carry out one-way ANOVA;
Look at how to examine contrasts (differences between the control or a reference factor level, and each of the others) and post-hoc testing (e.g., all pairwise comparisons between factor levels);
Examine what happens in the ANOVA in a little more detail.
ANOVA is not quite as simple in R as one might hope. Doing ANOVA takes at least two steps. First, we fit the ANOVA model to the data using the function lm(). This step carries out a bunch of intermediate calculations. Second, we use the results of first step to do the ANOVA calculations and place them in an ANOVA table using the function anova(). The function name lm() stands for “linear model”; this is actually a very powerful function that allows a variety of calculations. One-way ANOVA is a type of linear model.
7.1 Basic output
The basic output of the aov() function is a table of results. The table of results is a little hard to read, but it contains all the information we need to interpret the results of the ANOVA. The table of results is called an ANOVA table:
# Fit the modelmodel <-aov(Weight ~ Sire, data = data_long)# View the ANOVA tablesummary(model)
Df Sum Sq Mean Sq F value Pr(>F)
Sire 4 17426 4356 1.872 0.137
Residuals 35 81442 2327
The output is formatted in a classic “ANOVA Table” style. There are 2 rows - one for the “main effect” of the sire factor, one for the residual error. The test statistic is the F value (1.87), the P-value column is named “Pr(>F)” (0.14), and there are 4 degrees of freedom for this test for the factor (the factor degrees of freedom is the number of factor levels minus 1 = 5 factor levels - 1 = 4; the residual degrees of freedom is the total number of observations minus the number of factor levels = 40 - 5 = 35).
Here we can see that the overall effect of sire does not significantly explain variation in weight (one-way ANOVA: F = 1.87, df = 4,35, P = 0.14).
7.2 Contrasts
The ANOVA table tells us that there is no significant effect of sire on offspring weight. But, we might want to know which sires are different from each other. To do this, we need to perform contrasts and post-hoc testing. Contrasts are comparisons between factor levels. Post-hoc tests are multiple comparisons between factor levels. We will look at two types of contrasts. For our experiment, let’s say that sire C is our reference level sire, against which we would like to statistically compare offspring weight for other sires.
# Make Sire C the reference leveldata_long$Sire <-relevel(data_long$Sire, ref="C")# Fit linear modelmodel_2 <-lm(formula = Weight ~ Sire, data = data_long)# View the ANOVA tablesummary(model_2)
Call:
lm(formula = Weight ~ Sire, data = data_long)
Residuals:
Min 1Q Median 3Q Max
-93.625 -29.312 -2.875 33.906 104.750
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 696.63 17.05 40.846 <2e-16 ***
SireA 18.38 24.12 0.762 0.451
SireB -31.50 24.12 -1.306 0.200
SireD -39.13 24.12 -1.622 0.114
SireE -13.38 24.12 -0.555 0.583
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 48.24 on 35 degrees of freedom
Multiple R-squared: 0.1763, Adjusted R-squared: 0.08211
F-statistic: 1.872 on 4 and 35 DF, p-value: 0.1373
Notice how the output format has changed. This is because the summary function (and lots of functions in fact) “behave differently” in response to the specific class() of object we pass to it (here an lm object; before an aov object). One big difference we see is the table of contrasts. Now, there is are 5 rows: one for the intercept coefficient (testing whether the “grand mean” of Weight is different to zero - NB this is not at all interesting for us), and for rows comparing each factor level to the reference mean for sire “C”.
Here, the Estimate column is an estimate of the AMOUNT of difference in weight relative to the reference mean weight for that sire. E.g., sire B offspring weight is estimated at -31.5 grams (the negative indicate less than) compared to offspring weight for sire C. However, this observed sample difference is not statistically significant (P = 0.20).
Notice the overall F value and p-value for the one-way are also present at the bottom of the output, which is exactly the same as that produced by the aov() function.
7.3 Post-hoc testing
Post-hoc tests are often interesting in research, and the one-way ANOVA is a good example, where an overall question of “is there a difference?” can be enhanced by asking whether there are particular or specific differences, say between pairs of means. The phrase post-poc implies that the sometimes these questions can be an afterthought. Thus, consideration should be given as to whether these specific questions NEED to be asked.
There are many ways to perform post-hoc tests in R. A one-way ANOVA can tell us that at least one group has a different mean from another group, but it does not inform us which group means are different from which other group means. A Tukey-Kramer test lets us test the null hypothesis of no difference between the population means for all pairs of groups. The Tukey-Kramer test (also known as a Tukey Honestly Significance Test, or Tukey’s HSD), is implemented in R in the function TukeyHSD(). We will use the results of an ANOVA done with lm() as above, that we stored in the variable model_2. To do a Tukey-Kramer test on these data, we need to first apply the function aov() to model_2, and then we need to apply the function TukeyHSD() to the result. We can do this in a single command:
The key part of this output is the table. It shows:
The difference between the means of groups;
The 95% confidence interval for the difference between the corresponding population means (“lwr” and “upr” correspond to the lower and upper bounds of that confidence interval for the difference in means);
The p-value from a test of the null hypothesis of no difference between the means (the column headed with “p adj”). In the case of the chicken weight data, p is greater than 0.05 in all pairs, and we therefore accept every null hypothesis.
The output is a table of results, which is a little hard to read. We can improve the statistical output by using the agricolae package, which is not loaded by default when you start R.
Study: model ~ "Sire"
HSD Test for Weight
Mean Square Error: 2326.921
Sire, means
Weight std r se Min Max Q25 Q50 Q75
A 715.000 39.34826 8 17.05477 675 793 690.00 702.0 726.00
B 665.125 46.67345 8 17.05477 592 732 627.75 681.5 691.75
C 696.625 59.62726 8 17.05477 603 763 669.75 709.0 739.50
D 657.500 40.06067 8 17.05477 600 718 637.50 663.0 675.00
E 683.250 52.41796 8 17.05477 611 788 656.00 676.0 696.75
Alpha: 0.05 ; DF Error: 35
Critical Value of Studentized Range: 4.065949
Minimun Significant Difference: 69.34383
Treatments with the same letter are not significantly different.
Weight groups
A 715.000 a
C 696.625 a
E 683.250 a
B 665.125 a
D 657.500 a
This gives us a much neater output with convenient grouping letters to indicate where differences occur - different letters indicate that the group means are different. We can also plot the post-hoc analysis:
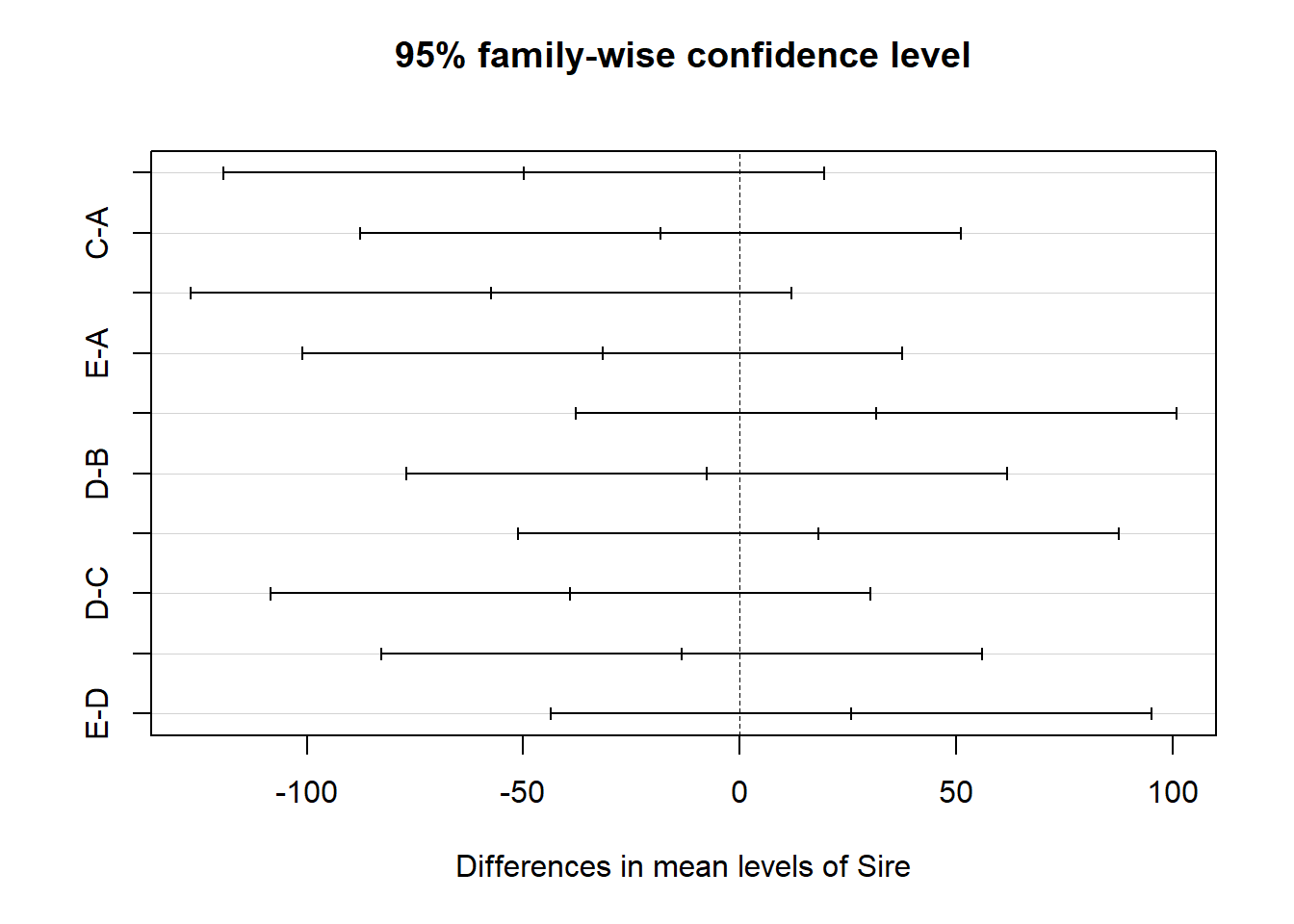
# Plot the resultsplot(TukeyHSD(model))
The plot shows the difference between each pair of means, with the 95% confidence intervals for each difference. The vertical line is the “null” difference of zero. If the confidence interval crosses the line, then the difference is not statistically significant. If the confidence interval does not cross the line, then the difference is statistically significant.
8 Two-way ANOVA
The two-way ANOVA is a statistical method that allows to evaluate the simultaneous effect of two categorical variables on a quantitative continuous variable. The two-way ANOVA is an extension of the one-way ANOVA since it allows to evaluate the effects on a numerical response of two categorical variables instead of one. The advantage of a two-way ANOVA over a one-way ANOVA is that we test the relationship between two variables, while taking into account the effect of a third variable. Moreover, it also allows to include the possible interaction of the two categorical variables on the response to evaluate whether or not they act jointly on the response variable.
8.1 Data
To illustrate how to perform a two-way ANOVA in R, we use the penguins data set, available from the palmerpenguins package. This data set contains information about 344 penguins from three species (Adelie, Chinstrap and Gentoo) collected from three islands in the Palmer Archipelago, Antarctica. We do not need to import the data set but we have to load the package then call it:
# Load the packagelibrary(palmerpenguins)
Warning: package 'palmerpenguins' was built under R version 4.3.2
# Call the datadat <- penguins # rename data setstr(dat) # structure of data set
tibble [344 × 8] (S3: tbl_df/tbl/data.frame)
$ species : Factor w/ 3 levels "Adelie","Chinstrap",..: 1 1 1 1 1 1 1 1 1 1 ...
$ island : Factor w/ 3 levels "Biscoe","Dream",..: 3 3 3 3 3 3 3 3 3 3 ...
$ bill_length_mm : num [1:344] 39.1 39.5 40.3 NA 36.7 39.3 38.9 39.2 34.1 42 ...
$ bill_depth_mm : num [1:344] 18.7 17.4 18 NA 19.3 20.6 17.8 19.6 18.1 20.2 ...
$ flipper_length_mm: int [1:344] 181 186 195 NA 193 190 181 195 193 190 ...
$ body_mass_g : int [1:344] 3750 3800 3250 NA 3450 3650 3625 4675 3475 4250 ...
$ sex : Factor w/ 2 levels "female","male": 2 1 1 NA 1 2 1 2 NA NA ...
$ year : int [1:344] 2007 2007 2007 2007 2007 2007 2007 2007 2007 2007 ...
The data set contains 344 rows and 8 columns. Each row corresponds to a penguin and each column corresponds to a variable. The variables we will focus on are:
species: the species of the penguin (Adelie, Chinstrap or Gentoo)
sex: sex of the penguin (female and male)
body_mass_g: body mass of the penguin (in grams)
body_mass_g is the quantitative continuous variable and will be the dependent variable, whereas species and sex are both qualitative variables. Those two last variables will be our independent variables, also referred as factors. Make sure that they are read as factors by R:
# Convert to factordat$species <-as.factor(dat$species)class(dat$species)
[1] "factor"
dat$sex <-as.factor(dat$sex)class(dat$sex)
[1] "factor"
8.2 Aim of the analysis
As mentioned above, a two-way ANOVA is used to evaluate simultaneously the effect of two categorical variables on one quantitative continuous variable. It is referred as two-way ANOVA because we are comparing groups which are formed by two independent categorical variables. Here, we would like to know if body mass depends on species and/or sex. In particular, we are interested in:
Measuring and testing the relationship between species and body mass,
Measuring and testing the relationship between sex and body mass, and
Potentially check whether the relationship between species and body mass is different for females and males (which is equivalent than checking whether the relationship between sex and body mass depends on the species)
The first two relationships are referred as main effects, while the third point is known as the interaction effect. The main effects test whether at least one group is different from another one (while controlling for the other independent variable). On the other hand, the interaction effect aims at testing whether the relationship between two variables differs depending on the level of a third variable. In other words, if the evolution between the response and the first categorical variable does not depend on the modalities of the second categorical variable, then there is no interaction between the two variables. If, on the contrary, there is a modification of this evolution, either by an increase in the effect of the first variable, or by a decrease, then there is an interaction.
When performing a two-way ANOVA, testing the interaction effect is not mandatory. However, omitting an interaction effect may lead to erroneous conclusions if the interaction effect is present.
8.3 Assumptions
Most statistical tests require some assumptions for the results to be valid, and a two-way ANOVA is not an exception.
Assumptions of a two-way ANOVA are similar than for a one-way ANOVA. To summarise:
Variable type: the dependent variable must be quantitative continuous, while the two independent variables must be categorical (with at least two levels).
Independence: the observations should be independent between groups and within each group.
Normality:
For small samples, data should follow approximately a normal distribution
For large samples (usually \(n \ge 30\) in each group/sample), normality is not required (thanks to the central limit theorem)
Equality of variances: variances should be equal across groups.
Outliers: There should be no significant outliers in any group.
Now that we have seen the underlying assumptions of the two-way ANOVA, we review them specifically for our data set before applying the test and interpreting the results.
8.3.1 Variable type
The dependent variable body mass is quantitative continuous, while both independent variables sex and species are qualitative variables (with at least 2 levels). Therefore, this assumption is met. If your dependent variable is quantitative discrete, this is count data, which, strictly speaking, should be analysed using a generalized linear model, not an ANOVA.
8.3.2 Independence
Independence is usually checked based on the design of the experiment and how data have been collected.1
To keep it simple, observations are usually:
independent if each experimental unit (here a penguin) has been measured only once and the observations are collected from a representative and randomly selected portion of the population, or
dependent if each experimental unit has been measured at least twice (as it is often the case in the medical field for example, with two measurements on the same subjects; one before and one after the treatment).
In our case, body mass has been measured only once on each penguin, and on a representative and random sample of the population, so the independence assumption is met.
Note that if your data correspond to observations made several times on the same experimental units (for example, if one of the factors is a treatment (A or B) and the second factor is time (day 1, day 2 and day 3), and measurements are made at each time point on the same subjects), a two-way mixed ANOVA should be used.
8.3.3 Normality
We have a large sample in all subgroups (each combination of the levels of the two factors, called cell):
For completeness, we still show how to verify normality, as if we had a small samples.
There are several methods to test the normality assumption. The most common methods being:
a QQ-plot by group or on the residuals;
a histogram by group or on the residuals;
a normality test by group or on the residuals.
The easiest/shortest way is to verify the normality with a QQ-plot on the residuals. To draw this plot, we first need to save the model:
# save modelmod <-aov(body_mass_g ~ sex * species,data = dat)
This piece of code will be explained further. Now we can draw the QQ-plot on the residuals. We show two ways to do so, first with the plot() function and second with the qqPlot() function from the {car} package:
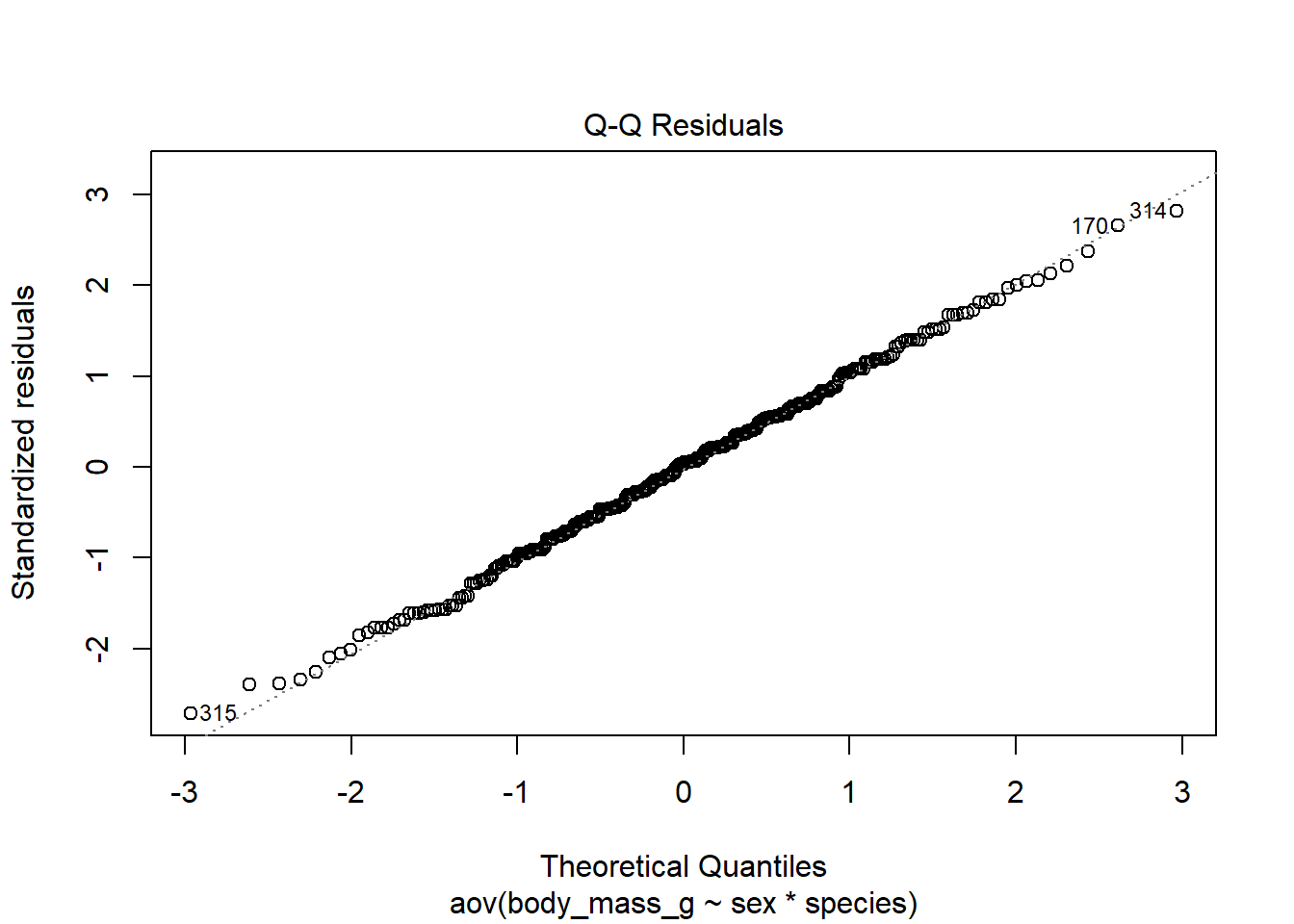
# method 1plot(mod, which =2)
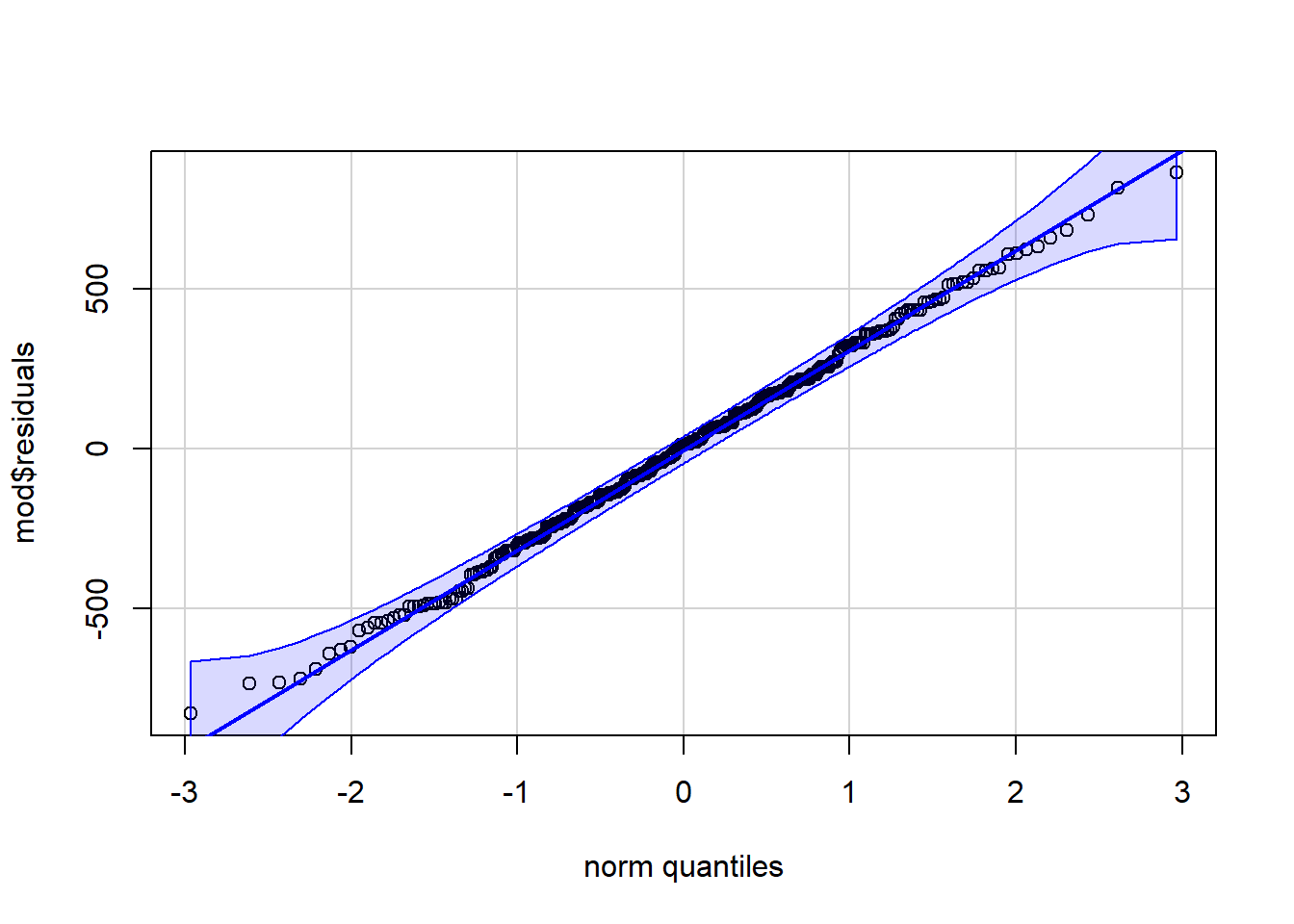
# method 2library(car)qqPlot(mod$residuals,id =FALSE# remove point identification)
Code for method 1 is slightly shorter, but it misses the confidence interval around the reference line.
If points follow the straight line and fall within the confidence band, we can assume normality. This is the case here. If you prefer to verify the normality based on a histogram of the residuals, here is the code:
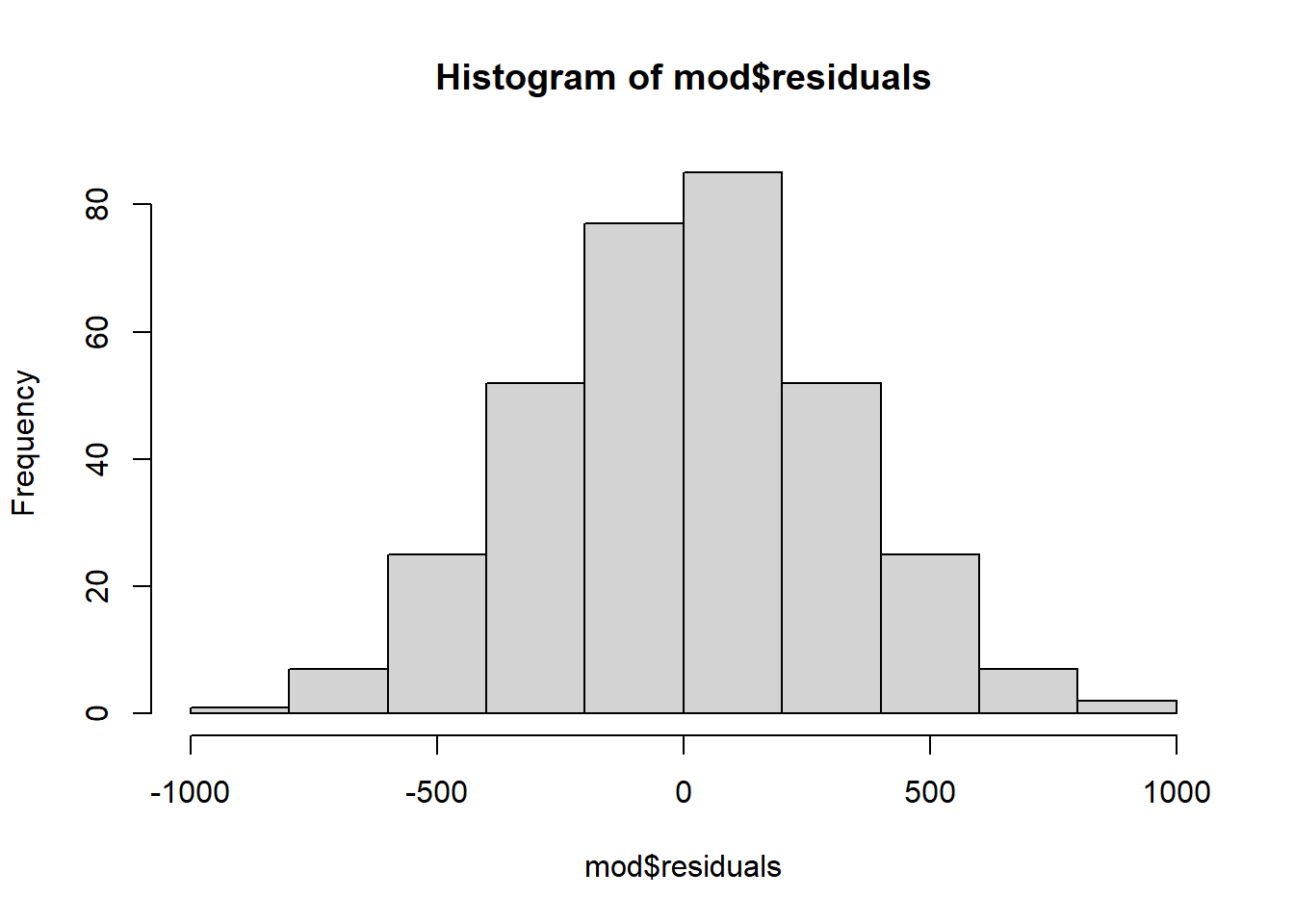
# histogramhist(mod$residuals)
The histogram of the residuals show a Gaussian distribution, which is in line with the conclusion from the QQ-plot. Although the QQ-plot and histogram is largely enough to verify the normality, if you want to test it more formally with a statistical test, the Shapiro-Wilk test can be applied on the residuals as well:
# normality testshapiro.test(mod$residuals)
Shapiro-Wilk normality test
data: mod$residuals
W = 0.99776, p-value = 0.9367
We do not reject the null hypothesis that the residuals follow a normal distribution (\(p\)-value = 0.937).
From the QQ-plot, histogram and Shapiro-Wilk test, we conclude that we do not reject the null hypothesis of normality of the residuals. The normality assumption is thus verified, we can now check the equality of the variances.
Note that if the normality assumption is not met, many transformations can be applied on the dependent variable to improve it, the most common ones being the logarithmic (log() function in R) and the Box-Cox transformations. If the normality assumption is still not met on the transformed data, the non-parametric version of the two-way ANOVA, the Scheirer–Ray–Hare test, can be used. Alternatively, a permutation test can also be used.
8.3.4 Homogeneity of variances
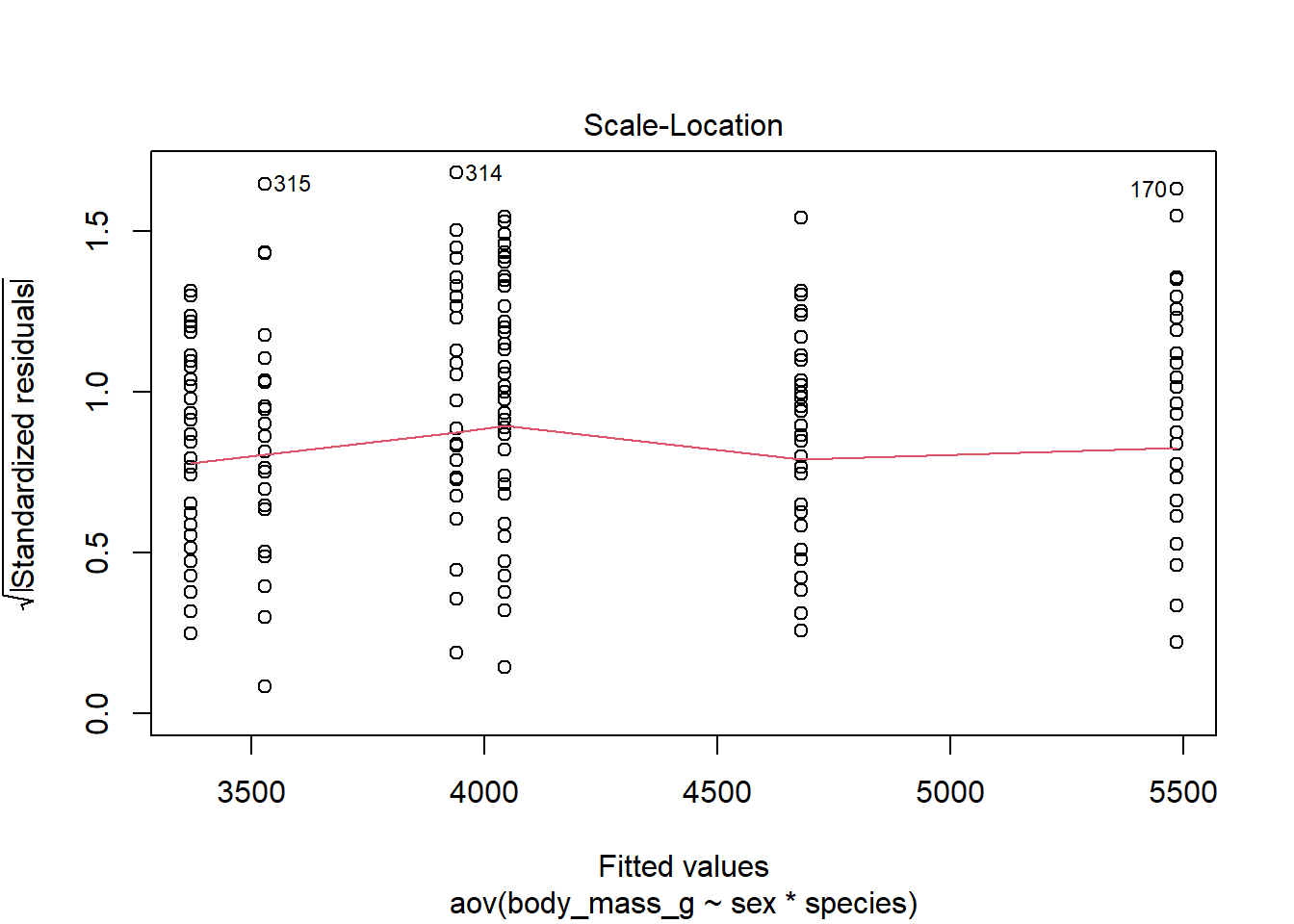
Equality of variances, also referred as homogeneity of variances or homoscedasticity, can be verified visually with the plot() function:
plot(mod, which =3)
Since the spread of the residuals is constant, the red smooth line is horizontal and flat, so it looks like the constant variance assumption is satisfied here.
The diagnostic plot above is sufficient, but if you prefer it can also be tested more formally with the Levene’s test (also from the {car} package):2
leveneTest(mod)
Levene's Test for Homogeneity of Variance (center = median)
Df F value Pr(>F)
group 5 1.3908 0.2272
327
We do not reject the null hypothesis that the variances are equal (\(p\)-value = 0.227).
Both the visual and formal approaches give the same conclusion; we do not reject the hypothesis of homogeneity of the variances.
Note that, as for the normality, the logarithmic and Box-Cox transformations may improve homogeneity of the residuals.
8.3.5 Outliers
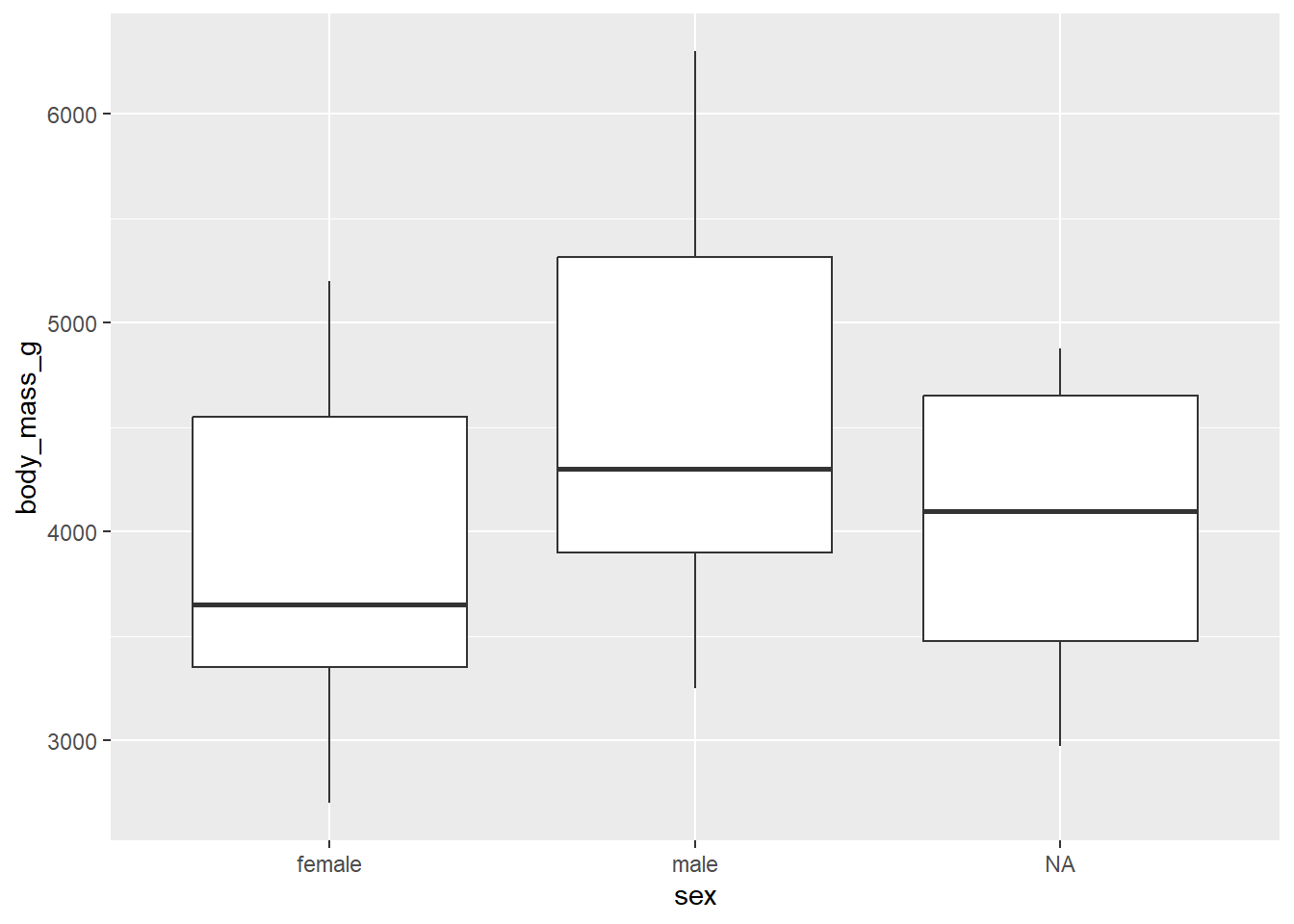
The easiest and most common way to detect outliersis visually thanks to boxplots by groups. For females and males:
library(ggplot2)# boxplots by sexggplot(dat) +aes(x = sex, y = body_mass_g) +geom_boxplot()
There are two outliers for the species Chinstrap. These points are, nonetheless, not extreme enough to bias results. Therefore, we consider that the assumption of no significant outliers is met.
8.4 Two-way ANOVA in R
We have shown that all assumptions are met, so we can now proceed to the implementation of the two-way ANOVA in R. This will allow us to answer the following research questions:
Controlling for the species, is body mass significantly different between the two sexes?
Controlling for the sex, is body mass significantly different for at least one species?
Is the relationship between species and body mass different between female and male penguins?
Including an interaction effect in a two-way ANOVA is not compulsory. However, in order to avoid flawed conclusions, it is recommended to first check whether the interaction is significant or not, and depending on the results, include it or not. If the interaction is not significant, it is safe to remove it from the final model. On the contrary, if the interaction is significant, it should be included in the final model which will be used to interpret results. We thus start with a model which includes the two main effects (i.e., sex and species) and the interaction:
# Two-way ANOVA with interaction# save modelmod <-aov(body_mass_g ~ sex * species,data = dat)# print resultssummary(mod)
Df Sum Sq Mean Sq F value Pr(>F)
sex 1 38878897 38878897 406.145 < 2e-16 ***
species 2 143401584 71700792 749.016 < 2e-16 ***
sex:species 2 1676557 838278 8.757 0.000197 ***
Residuals 327 31302628 95727
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
11 observations deleted due to missingness
Similar to a one-way ANOVA, the principle of a two-way ANOVA is based on the total dispersion of the data, and its decomposition into four components:
the share attributable to the first factor
the share attributable to the second factor
the share attributable to the interaction of the 2 factors
the unexplained, or residual portion.
The sum of squares (column Sum Sq) shows these four components. The two-way ANOVA consists of using a statistical test to determine whether each of the dispersion component (attributable to the 2 factors studied and to their interaction) is significantly greater than the residual component. If this is the case, we conclude that the effect considered (factor A, factor B or the interaction) is significant.
We see that the species explain a large part of the variability of body mass. It is the most important factor in explaining this variability.
The \(p\)-values are displayed in the last column of the output above (Pr(>F)). From these \(p\)-values, we conclude that, at the 5% significance level:
controlling for the species, body mass is significantly different between the two sexes;
controlling for the sex, body mass is significantly different for at least one species;
the interaction between sex and species (displayed at the line sex:species in the output above) is significant.
So from the significant interaction effect, we have just seen that the relationship between body mass and species is different between males and females. Since it is significant, we have to keep it in the model and we should interpret results from that model.
If, on the contrary, the interaction was not significant (that is, if the \(p\)-value \(\ge\) 0.05) we would have removed this interaction effect from the model. For illustrative purposes, below the code for a two-way ANOVA without interaction, referred as an additive model:
# Two-way ANOVA without interactionaov(body_mass_g ~ sex + species,data = dat)
8.5 Pairwise comparisons
Through the two main effects being significant, we concluded that:
controlling for the species, body mass is different between females and males;
controlling for the sex, body mass is different for at least one species.
If body mass is different between the two sexes, given that there are exactly two sexes, it must be because body mass is significantly different between females and males. If one wants to know which sex has the highest body mass, it can be deduced from the means and/or boxplots by subgroup. Here, it is clear that males have a significantly higher body mass than females.
However, it is not so straightforward for the species. Let me explain why it is not as easy as for the sexes.
There are three species (Adelie, Chinstrap and Gentoo), so there are 3 pairs of species:
Adelie and Chinstrap
Adelie and Gentoo
Chinstrap and Gentoo
If body mass is significantly different for at least one species, it could be that:
body mass is significantly different between Adelie and Chinstrap but not significantly different between Adelie and Gentoo, and not significantly different between Chinstrap and Gentoo, or
body mass is significantly different between Adelie and Gentoo but not significantly different between Adelie and Chinstrap, and not significantly different between Chinstrap and Gentoo, or
body mass is significantly different between Chinstrap and Gentoo but not significantly different between Adelie and Chinstrap, and not significantly different between Adelie and Gentoo.
Or, it could also be that:
body mass is significantly different between Adelie and Chinstrap, and between Adelie and Gentoo, but not significantly different between Chinstrap and Gentoo, or
body mass is significantly different between Adelie and Chinstrap, and between Chinstrap and Gentoo, but not significantly different between Adelie and Gentoo, or
body mass is significantly different between Chinstrap and Gentoo, and between Adelie and Gentoo, but not significantly different between Adelie and Chinstrap.
Last, it could also be that body mass is significantly different between all species. We cannot, at this stage, know precisely which species is different from which one in terms of body mass. To know this, we need to compare each species two by two thanks to post-hoc tests (also known as pairwise comparisons). There are several post-hoc tests, the most common ones being the Tukey HSD which tests all possible pairs of groups, and the Dunett’s test which compares all groups to a reference group. As mentioned earlier, these tests should not be done on the sex variable because there are only two levels. In this lab we will only look at Tukey HSD.
As for the one-way ANOVA, the Tukey HSD test can be done in R as follows:
# method 1TukeyHSD(mod,which ="species")
Tukey multiple comparisons of means
95% family-wise confidence level
Fit: aov(formula = body_mass_g ~ sex * species, data = dat)
$species
diff lwr upr p adj
Chinstrap-Adelie 26.92385 -80.0258 133.8735 0.8241288
Gentoo-Adelie 1377.65816 1287.6926 1467.6237 0.0000000
Gentoo-Chinstrap 1350.73431 1239.9964 1461.4722 0.0000000
or using the {multcomp} package:
# method 2library(multcomp)
Warning: package 'multcomp' was built under R version 4.3.2
Loading required package: mvtnorm
Warning: package 'mvtnorm' was built under R version 4.3.2
Loading required package: survival
Loading required package: TH.data
Warning: package 'TH.data' was built under R version 4.3.2
Loading required package: MASS
Attaching package: 'TH.data'
The following object is masked from 'package:MASS':
geyser
res_tukey <-glht(aov(body_mass_g ~ sex + species,data = dat ),linfct =mcp(species ="Tukey"))summary(res_tukey)
Simultaneous Tests for General Linear Hypotheses
Multiple Comparisons of Means: Tukey Contrasts
Fit: aov(formula = body_mass_g ~ sex + species, data = dat)
Linear Hypotheses:
Estimate Std. Error t value Pr(>|t|)
Chinstrap - Adelie == 0 26.92 46.48 0.579 0.83
Gentoo - Adelie == 0 1377.86 39.10 35.236 <1e-05 ***
Gentoo - Chinstrap == 0 1350.93 48.13 28.067 <1e-05 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Adjusted p values reported -- single-step method)
Note that when using the second method, it is the model without the interaction that needs to be specified into the glht() function, even if the interaction is significant. Moreover, do not forget to replace mod and species in my code with the name of your model and the name of your independent variable.
Both methods give the same results, that is:
body mass is not significantly different between Chinstrap and Adelie (adjusted \(p\)-value = 0.83),
body mass is significantly different between Gentoo and Adelie (adjusted \(p\)-value < 0.001), and
body mass is significantly different between Gentoo and Chinstrap (adjusted \(p\)-value < 0.001).
Remember that it is the adjusted\(p\)-values that are reported, to prevent the issue of multiple testing which occurs when comparing several pairs of groups.
If you would like to compare all combinations of groups, it can be done with the TukeyHSD() function and specifying the interaction in the which argument:
# all combinations of sex and speciesTukeyHSD(mod,which ="sex:species")
Or with the HSD.test() function from the {agricolae} package, which denotes subgroups that are not significantly different from each other with the same letter:
Study: mod ~ c("sex", "species")
HSD Test for body_mass_g
Mean Square Error: 95726.69
sex:species, means
body_mass_g std r se Min Max Q25 Q50
female:Adelie 3368.836 269.3801 73 36.21222 2850 3900 3175.00 3400
female:Chinstrap 3527.206 285.3339 34 53.06120 2700 4150 3362.50 3550
female:Gentoo 4679.741 281.5783 58 40.62586 3950 5200 4462.50 4700
male:Adelie 4043.493 346.8116 73 36.21222 3325 4775 3800.00 4000
male:Chinstrap 3938.971 362.1376 34 53.06120 3250 4800 3731.25 3950
male:Gentoo 5484.836 313.1586 61 39.61427 4750 6300 5300.00 5500
Q75
female:Adelie 3550.00
female:Chinstrap 3693.75
female:Gentoo 4875.00
male:Adelie 4300.00
male:Chinstrap 4100.00
male:Gentoo 5700.00
Alpha: 0.05 ; DF Error: 327
Critical Value of Studentized Range: 4.054126
Groups according to probability of means differences and alpha level( 0.05 )
Treatments with the same letter are not significantly different.
body_mass_g groups
male:Gentoo 5484.836 a
female:Gentoo 4679.741 b
male:Adelie 4043.493 c
male:Chinstrap 3938.971 c
female:Chinstrap 3527.206 d
female:Adelie 3368.836 d
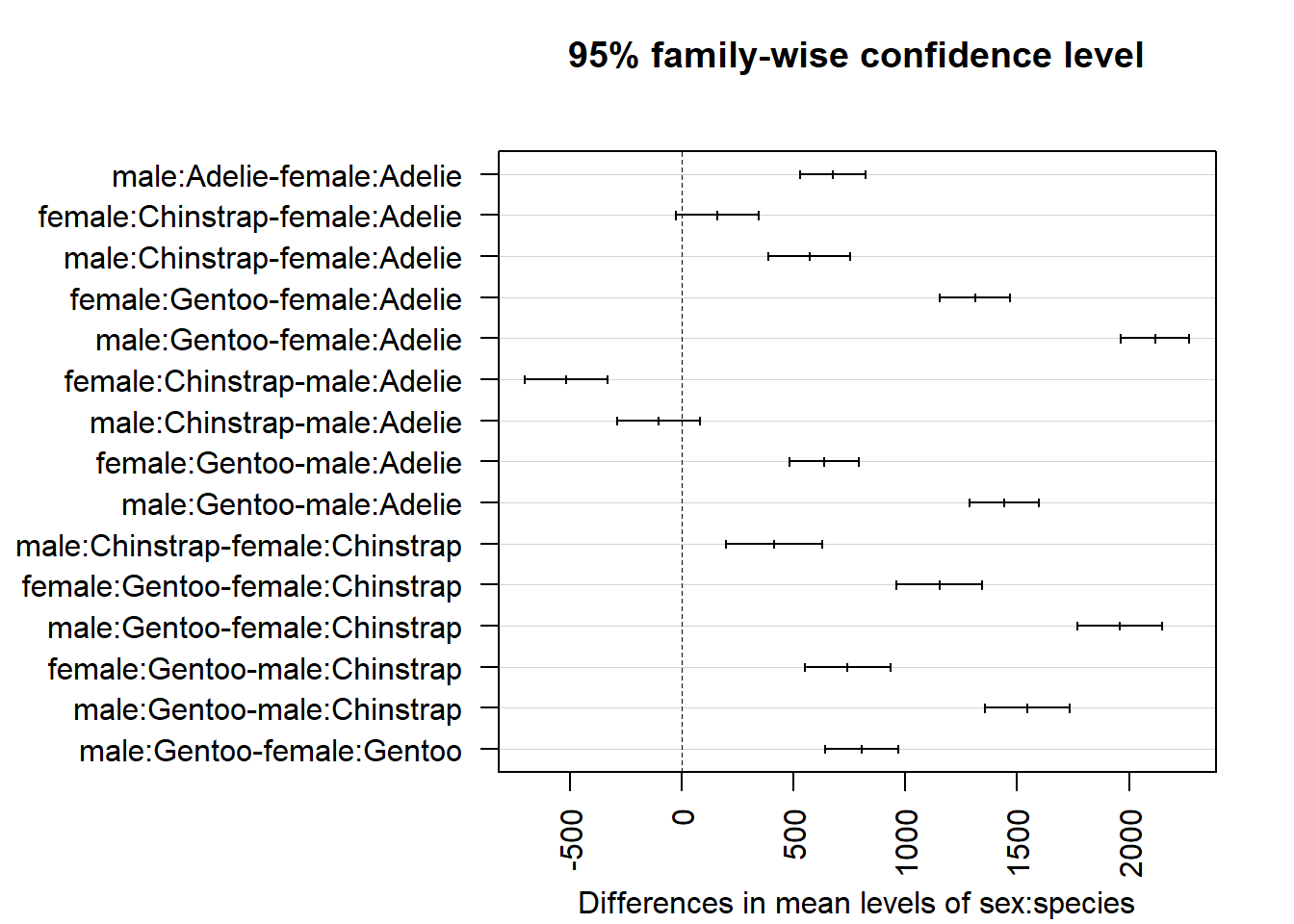
If you have many groups to compare, plotting them might be easier to interpret:
# set axis margins so labels do not get cut offpar(mar=c(4.1, 13.5, 4.1, 2.1))# create confidence interval for each comparisonplot(TukeyHSD(mod, which ="sex:species"),las =2# rotate x-axis ticks )
From the outputs and plot above, we conclude that all combinations of sex and species are significantly different, except between female Chinstrap and female Adelie (\(p\)-value = 0.138) and male Chinstrap and male Adelie (\(p\)-value = 0.581).
9 Non-parametric alternative
A Kruskal-Wallis test is a non-parametric analog of a one-way ANOVA. It does not assume that the numeric variable has a Gaussian distribution for each group. It is implemented in R in the function kruskal.test(). The Kruskal-Wallis test is a rank-based test, and it is therefore not affected by outliers in the same way as a one-way ANOVA. The Kruskal-Wallis test is also more robust to unequal variances than a one-way ANOVA. However, the Kruskal-Wallis test is less powerful than a one-way ANOVA when the data do in fact come from Gaussian distributions with equal variances. The input for this function is the same as we used for lm() above. It includes a model formula statement and the name of the data frame to be used:
# Perform Kruskal-Wallis testkruskal.test(Weight ~ Sire, data = data_long)
Kruskal-Wallis rank sum test
data: Weight by Sire
Kruskal-Wallis chi-squared = 7.648, df = 4, p-value = 0.1054
You can see for the output that a Kruskal-Wallis test also confirms that we can accept the null hypothesis of no difference between the means of the groups (p = 0.105).
10 Transforming Data
Up until now, the data we’ve examined have conformed, at least roughly, to the assumptions of the statistical models we’ve been studying. This is all very handy, but perhaps a little unrealistic. The real world being the messy place it is, biological data often don’t conform to the distributional assumptions of t-tests and ANOVA:
The residuals may not be normally distribute;
Variances may be unequal among groups.
The same kinds of problems with these distributional assumptions can also arise when working in a regression (i.e. non-normal residuals, non-constant variance). Furthermore, we might run into additional problems if there is some kind of non-linearity in the relationship between the response and predictor (numeric) variables.
Most biological data are unlikely to conform perfectly to all the assumptions, but experience has shown (fortunately) that t-tests, ANOVAs and regressions are generally quite robust—they perform reasonably well with data that deviate to some extent from the assumptions of the tests. However, in some cases residuals are clearly very far from normal, or variances change a lot across groups.
In these cases steps may need to be taken to deal with the problem. This chapter deals with one way of tackling the analysis of data that don’t fit the assumptions: data transformation. We will mostly focus on ANOVA / t-test setting, but keep in mind that the ideas are equally applicable to regression analysis.
10.1 Log transformations
Log transformation affects the data in two ways:
A log-transformation stretches out the left hand side (smaller values) of the distribution and squashes in the right hand side (larger values). This is obviously useful where the data set has a long tail to the right as in the example above;
The ‘squashing’ effect of a log-transformation is more pronounced at higher values. This means a log-transformation may also deal with another common problem in biological data (also seen in the ant data)—samples with larger means having larger variances.
If we are carrying out an ANOVA and the scale-location plot exhibits a positive relationship—i.e., groups with larger means have larger variances—then a log transformation could be appropriate. You can log transform the data using the log() function in R. For example, to log transform the Weight variable in the chicken data, you would use:
# Log transform the datadata_long$logWeight <-log(data_long$Weight)
10.2 Square root transformations
Taking the square root of the data is often appropriate where the data are whole number counts (though the log transformation may also work here). This typically occurs where your data are counts of organisms (e.g., algal cells in fields of view under a microscope). In R the square root of a set of data can be taken using the sqrt() function. However, note that there is no square function in the list. Taking squares is done using the ^ operator with the number 2 on the right (e.g., if the variable is called x, use x^2). To this with our chicken data, we would use:
# Square root transform the datadata_long$sqrtWeight <-sqrt(data_long$Weight)
10.3 Not all data can be transformed
There are some situations where no amount of transformation of the data will get round the fact that the data are problematic. Three in particular are worth noting…
Multimodal distributions - these may in fact have only one actual mode, but nonetheless have two or more clear ‘peaks’ (N.B. not to be confused with distributions that are ‘spiky’ just because there are few data);
Dissimilar distribution shapes - if the two (or more) samples have very different problems, e.g. one is strongly right-skewed and the other strongly left-skewed then no single transformation will be able to help—whatever corrects one sample will distort the other;
Samples with many exactly equal values - with results that are small integer numbers (e.g. counts of numbers of eggs in birds’ nests) then there will be many identical values. If the non-normality results from lots of identical values forming a particular peak, for example, this cannot be corrected by transformation since equal values will still be equal even when transformed.
11 Activities
11.1 Cuckoo eggs
The European cuckoo does not look after its own eggs, but instead lays them in the nests of birds of other species. Previous studies showed that cuckoos sometimes have evolved to lay eggs that are colored similarly to the host bird species. Is the same true of egg size? Do cuckoos lay eggs similar in size to the size of the eggs of their hosts? The data file cuckooeggs.csv contains data on the lengths of cuckoo eggs laid in the nests of a variety of host species. Here we compare the mean size of cuckoo eggs found in the nests of different host species. These are your tasks:
Read the data into R and check the data for errors;
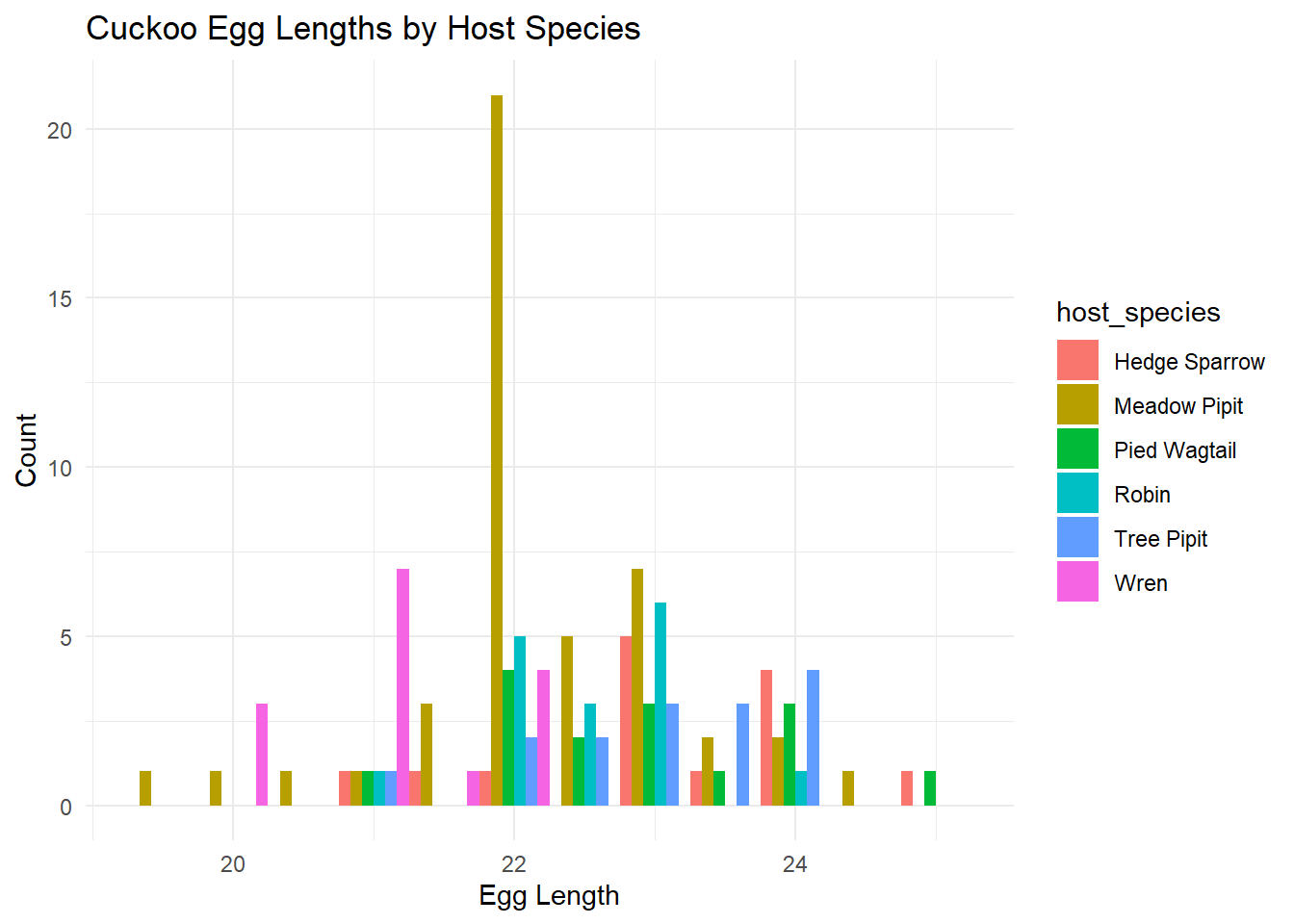
Plot a multiple histogram showing cuckoo egg lengths by host species;
Calculate a table that shows the mean and standard deviation of length of cuckoo eggs for each host species;
Look at the graph and the table. For these data, would ANOVA be a valid method to test for differences between host species in the lengths of cuckoo eggs in their nests?
Use ANOVA to test for a difference between host species in the mean size of the cuckoo eggs in their nests. What is your conclusion?
Assuming that ANOVA rejected the null hypotheses of no mean differences, use a Tukey-Kramer test to decide which pairs of host species are significantly different from each other in cuckoo egg mean length. What is your conclusion?
# Plotting a multiple histogramggplot(cuckoo_data, aes(x = egg_length, fill = host_species)) +geom_histogram(position ="dodge", binwidth =0.5) +theme_minimal() +labs(title ="Cuckoo Egg Lengths by Host Species", x ="Egg Length", y ="Count")
# Calculate mean and standard deviation by host speciessummary_table <- cuckoo_data %>%group_by(host_species) %>%summarise(mean_length =mean(egg_length),sd_length =sd(egg_length))# Print the summary tableprint(summary_table)
# Tukey HSD test with agricolaeHSD.test(anova_result, "host_species", console =TRUE)
Study: anova_result ~ "host_species"
HSD Test for egg_length
Mean Square Error: 0.8267399
host_species, means
egg_length std r se Min Max Q25 Q50 Q75
Hedge Sparrow 23.12143 1.0687365 14 0.2430079 20.85 25.05 22.90 23.05 23.85
Meadow Pipit 22.29889 0.9206278 45 0.1355433 19.65 24.45 22.05 22.25 22.85
Pied Wagtail 22.90333 1.0676186 15 0.2347680 21.05 24.85 21.95 23.05 23.75
Robin 22.57500 0.6845923 16 0.2273131 21.05 23.85 22.05 22.55 23.05
Tree Pipit 23.09000 0.9014274 15 0.2347680 21.05 24.05 22.55 23.25 23.75
Wren 21.13000 0.7437357 15 0.2347680 19.85 22.25 20.85 21.05 21.75
Alpha: 0.05 ; DF Error: 114
Critical Value of Studentized Range: 4.099489
Groups according to probability of means differences and alpha level( 0.05 )
Treatments with the same letter are not significantly different.
egg_length groups
Hedge Sparrow 23.12143 a
Tree Pipit 23.09000 a
Pied Wagtail 22.90333 ab
Robin 22.57500 ab
Meadow Pipit 22.29889 b
Wren 21.13000 c
11.2 Malaria
The pollen of the maize (corn) plant is a source of food to larval mosquitoes of the species Anopheles arabiensis, the main vector of malaria in Ethiopia. The production of maize has increased substantially in certain areas of Ethiopia recently, and over the same time period, malaria has entered in to new areas where it was previously rare. This raises the question, is the increase of maize cultivation partly responsible for the increase in malaria?
One line of evidence is to look for an association between maize production and malaria incidence at different geographically dispersed sites. The data set malaria vs maize.csv contains information on several high-altitude sites in Ethiopia, with information about the level of cultivation of maize (low, medium or high in the variable maize_yield) and the rate of malaria per 10,000 people (incidence_rate_per_ten_thousand). These are your tasks:
Read the data into R and check the data for errors;
Plot a multiple histogram to show the relationship between level of maize production and the incidence of malaria;
ANOVA is a logical choice of method to test differences in the mean rate of malaria between sites differing in level of maize production. Calculate the standard deviation of the incidence rate for each level of maize yield. Do these data seem to conform to the assumptions of ANOVA? Describe any violations of assumptions you identify;
Compute the log of the incidence rate and redraw the multiple histograms for different levels of maize yield. Calculate the standard deviation of the log incidence rate for each level of maize yield. Does the log-transformed data better meet the assumptions of ANOVA than did the untransformed data?
Test for an association between maize yield and malaria incidence.
💡 Click here to view a solution
library(tidyverse)library(agricolae)# Read the datadata <-read.csv("data/malaria_maize.csv")# Plot multiple histogramsggplot(data, aes(x = incidence_rate_per_ten_thousand, fill = maize_yield)) +geom_histogram(position ="dodge", binwidth =10) +labs(title ="Histogram of Malaria Incidence Rate by Maize Yield Level",x ="Incidence Rate per Ten Thousand",y ="Frequency",fill ="Maize Yield Level")# Standard Deviation of Incidence Rate by Maize Yield Levelstd_dev_by_yield <- data %>%group_by(maize_yield) %>%summarize(StdDev_Incidence_Rate =sd(incidence_rate_per_ten_thousand))print(std_dev_by_yield)# Checking for ANOVA assumptions: Homogeneity of variances# (Bartlett's test is commonly used for this)bartlett_test <-bartlett.test(incidence_rate_per_ten_thousand ~ maize_yield, data = data)print(bartlett_test)# Log Transformation and Redrawing Histogramsdata$log_incidence_rate <-log(data$incidence_rate_per_ten_thousand)ggplot(data, aes(x = log_incidence_rate, fill = maize_yield)) +geom_histogram(position ="dodge", binwidth =0.5) +labs(title ="Histogram of Log-Transformed Malaria Incidence Rate by Maize Yield Level",x ="Log of Incidence Rate",y ="Frequency",fill ="Maize Yield Level")# Standard Deviation of Log Incidence Rate by Maize Yield Levelstd_dev_log_by_yield <- data %>%group_by(maize_yield) %>%summarize(StdDev_Log_Incidence_Rate =sd(log_incidence_rate))print(std_dev_log_by_yield)# 4. Test for Association# Using ANOVA to test for differences in means between groupsanova_result <-aov(incidence_rate_per_ten_thousand ~ maize_yield, data = data)summary(anova_result)# Using Tukey HSD from agricolaeHSD.test(anova_result, "maize_yield", console =TRUE)
Footnotes
If you really want to test the independence, you can do so visually with a plot of the residuals vs. fitted values. This plot can be done in R with plot(mod, which = 1), where mod corresponds to the name of your model. Or you can do so with the Durbin-Watson test. In R, it can be done with the durbinWatsonTest() function from the car package.↩︎
Note that the Bartlett’s and Fligner-Killeen tests are also appropriate to test the assumption of equal variances.↩︎